
Abstract
Background
As a degenerative joint disease, osteoarthritis (OA) is characterized by articular cartilage degradation. Long noncoding RNAs (lncRNAs) act critical roles in the regulation of OA development, including affecting the proliferation, apoptosis, extracellular matrix (ECM) degradation, and inflammatory response of chondrocytes. The current study’s aim was to investigate the regulatory function and the underlying molecular mechanism of lncRNA MEG3 in ECM degradation of chondrocytes in OA.
Methods
In the current study, chondrocytes were induced by interleukin-1β (IL-1β) to simulate OA condition, and further assessed cell viability, lncRNA MEG3 and miR-93 expression levels. Overexpression or knockdown of lncRNA MEG3 in chondrocytes treated with IL-1β were performed to investigate the function of MEG3 in regulating cell proliferation, apoptosis and ECM degradation using EdU assay, flow cytometry, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and Western blot. The interaction between MEG3 and miR-93 was assessed using qRT-PCR. Furthermore, overexpression of miR-93 was performed as recovery experiment to explore the functional mechanism of MEG3.
Results
MEG3 was significantly downregulated in chondrocytes treated with IL-1β, whereas miR-93 was upregulated concomitantly. Overexpression of MEG3 induced the proliferation, suppressed the apoptosis, and relieved the degradation of ECM in IL-1β-induced chondrocytes. By contrast, knockdown of MEG3 suppressed the proliferation, promoted the apoptosis, and aggravated ECM degradation in IL-1β induced chondrocytes. In addition, MEG3 was found to relieve the inhibitive expression of TGFBR2 as a competitive endogenous RNA (ceRNA) of miR-93, and then activated transforming growth factor-β (TGF-β) signaling pathway, regulated chondrocytes ECM degradation in IL-1β induced chondrocytes subsequently.
Conclusion
LncRNA MEG3 targeted miR-93/TGFBR2 axis, regulated the proliferation, apoptosis and ECM degradation of chondrocytes in OA.
Introduction
Osteoarthritis (OA) is the most common form of arthritis. According to one recent report, half of individuals older than 65 years are affected by OA. 1 With the changing of lifestyle and the aging of population, degenerative joint disease has turned into a worldwide major cause of pain, physical debilitation, and shortening of working age. 2 As a typical degenerative joint disease, OA is characterized by articular cartilage degradation. 3 The imbalance between the synthesis and the degradation of extracellular matrix (ECM) of chondrocytes plays an important role in functional incapacitation and destruction of articular cartilage, which further lead to OA. 4 Previous studies showed that the metabolism of chondrocytes ECM is regulated by cytokines, degrading enzymes, and growth factors, some of which have been proved to relieve or reverse OA in vitro and in vivo.5,6 Most of the current treatments of OA focus on symptom control and pain management, such as glucocorticoids, analgesics, and nonsteroidal anti-inflammatory drugs (NSAIDs). But they are unable to prevent the degradation of chondrocytes ECM or the functional incapacitation of articular cartilage. 7 Therefore, the exploration of effective treatment against ECM degradation is crucial.
Long noncoding RNAs (lncRNAs) are nonprotein coding RNA transcripts with length above 200 bp. 8 LncRNAs were reported to be important regulators in several processes of the development and progression of OA, including the proliferation, apoptosis, ECM degradation, and inflammatory response of chondrocytes.9-12 In a rat model, downregulated maternally expressed gene 3 (MEG3) was observed in OA cartilage tissues and considered to be correlated with the alleviation of OA development, given that MEG3 regulated the proliferation and the apoptosis of chondrocytes induced by inerleukin-1β (IL-1β). 12 Meanwhile, the upregulation of MEG3 was reported to reduce OA attendant inflammation and pain by inhibiting angiogenesis. 11 MEG3 was also reported to act as a competitive endogenous RNA (ceRNA) of several miRNAs. miR-125a-5p 13 and miR-770-5p 14 were reported to have interactions with MEG3 in the researches of immune thrombocytopenic purpura and Hirschsprung’s disease. In gliomas, Zhang et al. 15 found that MEG3 inhibited cell growth via absorbing miR-93, which was reported to be upregulated in OA.16,17 In addition, as a direct target of miR-93, transforming growth factor β receptor type II (TGFBR2) was also involved in the progress of OA. 18 Knock-out of TGFBR2 resulted in obvious OA-like phenotype in mouse model. Downregulation of TGFBR2 induced the expression levels of runt-related transcription factor 2 (RUNX2), matrix metallopeptidase 13 (MMP-13), and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS-5), which could lead to a progressive OA development. 19 Nevertheless, the functional role of MEG3 in ECM degradation and the underlying mechanism in OA development remains unclear.
In this study, we raised the hypothesis that lncRNA MEG3 targets miR-93/TGFBR2 axis, suppresses the degradation of chondrocytes ECM and further recedes OA progression.
Materials and Methods
Experimental Animals
Male Sprague-Dawley rats (SD rats, 4 weeks) were used in this study. All rats were treated according to the national guidelines of the care and use of laboratory animals with the approval of the Ethics Committee for Animal Research.
Cell Culture and IL-1β Stimulation
SD rats were sacrificed under anesthesia. The articular cartilages of knee joints were separated under sterile condition and washed for 3 times with Dulbecco’s modified Eagle’s medium (DMEM). The attached muscle, perichondrium, and connective tissue were removed. The remaining articular cartilage samples were cut up, washed 3 times with DMEM, and trypsinized with 0.25% trypsin (Gibco; Carlsbad, CA, USA) for 30 minutes at 37°C, then incubated with 0.2% collagenase II (Invitrogen, Carlsbad, CA, USA) in DMEM for 6 hours. The samples were stirred every 30 minutes. After 2 hours, undissociated tissue was filtrated by 40 mm filter. Chondrocytes were isolated using centrifugation and grown at 37°C with 5% CO2 in DMEM, which was supplemented with 10% fetal bovine serum (Hyclone; Logan, UT, USA), 100 units/mL of streptomycin (Gibco; Carlsbad, CA, USA), and 100 units/mL of penicillin (Gibco; Carlsbad, CA, USA). Chondrocytes within 2 or 3 passage were used in this study. Chondrocytes were plated in 6-well plates at 5 × 105/well. When 70% to 80% confluence was met, chondrocytes were cultured in serum-free medium for 12 hours, and then stimulated by IL-1β for 48 hours.
Cell Transfection and IL-1β Stimulation
Green fluorescent protein (GFP) vector was used to estimate transfection efficiency. Full length of lncRNA MEG3 was inserted into pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) using the multiple cloning site. The correct construct of MEG3 overexpression vector were confirmed using DNA sequencing. Small interfering RNA of MEG3 (si-MEG3) and miR-93 mimic/inhibitor were ordered from RiboBio (Guangzhou, China). Chondrocytes were plated into 6-well plates 24 hours before transfection. Vectors and miRNAs were transfected with Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Forty-eight hours after the transfection, chondrocytes were then induced by IL-1β (10 ng/mL) for 48 hours.
MTT Assay
Chondrocytes were seeded into 96-well culture plates at 2,000/well. Twenty microliters of methylthiazolyldiphenyl-tetrazolium bromide (MTT) reagent (5 mg/mL; Sigma-Aldrich; St. Louis, MO, USA) was added into each well after treatment and then incubated chondrocytes for 4 hours at 37°C. After the incubation, the supernatant was removed, and the formazan crystals were dissolved using dimethylsulfoxide (DMSO, Sigma-Aldrich; St. Louis, MO, USA) (150 μL/well). The absorbance at 490 nm of each well was assessed by Microplate System. Experiments were conducted in triplicate.
EdU Assay
Chondrocytes proliferation was assessed by 5′-ethynyl-29-deoxyuridine (EdU) assay using Click-iT EdU Imaging Kits (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction. In brief, chondrocytes were plated in 96-well plate at 3,000/well. After treatment, cells were exposed to EdU (20 μM) at 37°C for 4 hours, fixed in 4% formaldehyde for 2 minutes, and then treated with 0.5% Triton X-100 at room temperature for 20 minutes. Cells were washed using 3% bull serum albumin (BSA) in phosphate buffered saline (PBS) twice. Wash solution was removed, reaction cocktail was added to each well (0.1 mL/well) and incubated at room temperature for 30 minutes. The reaction cocktail was then removed, and cells were washed with 3% BSA in PBS. Cells were incubated with Hoechst solution (1:2,000) at room temperature in dark for 30 minutes. After incubation, the Hoechst was removed and cells were washed with PBS, then observed and photographed using fluorescent microscope (Olympus, Tokyo, Japan). Image-Pro Plus (Media Cybernetics, Bethesda, MD, USA) was used to analyze the EdU incorporation rate as the ratio of EdU-positive cells (red) to total Hoechst-positive cells (blue).
Cell Apoptosis Assay
The apoptosis of chondrocytes was analyzed by the flow cytometry with Annexin V–FITC apoptosis detection kit (Sigma-Aldrich; St. Louis, MO, USA), following the instructions of manufacturer. In brief, chondrocytes were trypsinized and washed with chilled PBS, then resuspended in the binding buffer. Five microliters of FITC-labeled annexin V reagent was added to cell suspension (190 μL each), mixed gently, then 5 μL of propidium iodide (PI) solution was added to each sample. Samples were incubated in dark at room temperature for 20 minutes, and then analyzed using flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed by the FlowJo (FlowJo LLC, Ashland, OR, USA) software. Necrotic and viable cells were presented by the upper and lower left quadrants, the late and early apoptotic cells were presented by the right quadrants represent, respectively. The apoptosis rate was identified as the sum of the early apoptosis rate and the late apoptosis rate. Experiments were conducted in triplicate.
RNA Isolation and qRT-PCR Assay
Total RNA was isolated using TRIzol reagent (Sigma-Aldrich; St. Louis, MO, USA), and then reverse-transcribed using random primer or miRNA reverse-transcription primer and PrimeScript RT reagent kit with gDNA Eraser (Takara). qRT-PCR (quantitative reverse transcription polymerase chain reaction) primers of mRNAs were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast). Reverse-transcription primer and qRT-PCR Primers of miRNAs were ordered from RiboBio (Guangzhou, China). The qRT-PCR cycling was performed at 98°C for 2 minutes, then 40 cycles at the conditions of 98°C/15 seconds and 60°C/40 seconds. The relative expression of target genes was calculated using the formula 2−∆∆Ct. Sample lacking cDNA was used as negative control. GAPDH and U6 were chosen as internal control for mRNA and miRNA identification, respectively.
Western Blot
Cells were harvested and washed with PBS, then lysed with lysis buffer (Sigma-Aldrich; St. Louis, MO, USA). Cell lysate was gathered after centrifuge, and the protein concentration was determined by the bicinchoninic acid (BCA) protein assay (Vigorous). Proteins were quantified using electrophoresis with Tris-glycine polyacrylamide gels. Resolved protein was transferred to nitrocellulose membranes, and then blocked in 2.5% BSA for 1 hour, incubated with the primary antibody to MMP-13, ADAMTS-5, COL2A1, TGFBR2 (Abcam, 1:1000 dilution) and β-actin (Cell Signaling, 1:4000 dilution) at 4°C overnight. After washed with PBS, the membranes were incubated with second antibody (ZSGB-Bio, 1:4000 dilution) for 1 hour. The bands of proteins were detected by a chemiluminescence detection system (CWBIO; Beijing, China). The optical density of the protein bands was quantified using Image J software and was used to calculate the relative expression of target protein. The expression of β-actin was used as an internal control.
Immunofluorescence Staining
Chondrocytes were cultured in 12-well plates. After treatment, chondrocytes were fixed in 4% paraformaldehyde for 20 minutes, incubated with 0.3% Triton X-100 for 10 minutes, and then blocked using 5% BSA for 1 hour. Subsequently, chondrocytes were incubated with primary antibodies (rabbit polyclonal antibody) of MMP-13 and COL2A1 (Abcam, Cambridge, UK; 1:100) at 4°C overnight. Cells were then washed with PBS and incubated with Alexa Fluor 488 (green)- or 594 (red)-conjugated goat anti-rabbit (Invitrogen, Carlsbad, CA, USA) at room temperature for 1 hour. DAPI (Sigma-Aldrich; St. Louis, MO, USA) was used to stain the nuclear. Images were captured by fluorescence microscope.
Statistical Analysis
Data were expressed as mean ± standard deviation (mean ± SD) of the mean of at least 3 replicates. For comparison among multiple groups, one-way analysis of variance followed by the Bonferroni post hoc test was conducted. For 2-group comparisons, unpaired Student t test was used. P < 0.05 was considered statistically significant for Student t test. All statistical analyses were conducted using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
IL-1β Induced MEG3 Downregulation and miR-93 Upregulation in Chondrocytes
To explore the regulatory function of lncRNA MEG3 and miR-93 in OA, we simulated OA condition using IL-1β, and assessed the expression levels of MEG3, miR-93, and OA-related factors in the chondrocytes treated with IL-1β. Using MTT assay, we assessed the cell viability of chondrocytes treated with IL-1β (0-15 ng/mL) for 48 hours and found that the viability of chondrocytes was significantly reduced by IL-1β stimulation in a concentration-dependent manner (
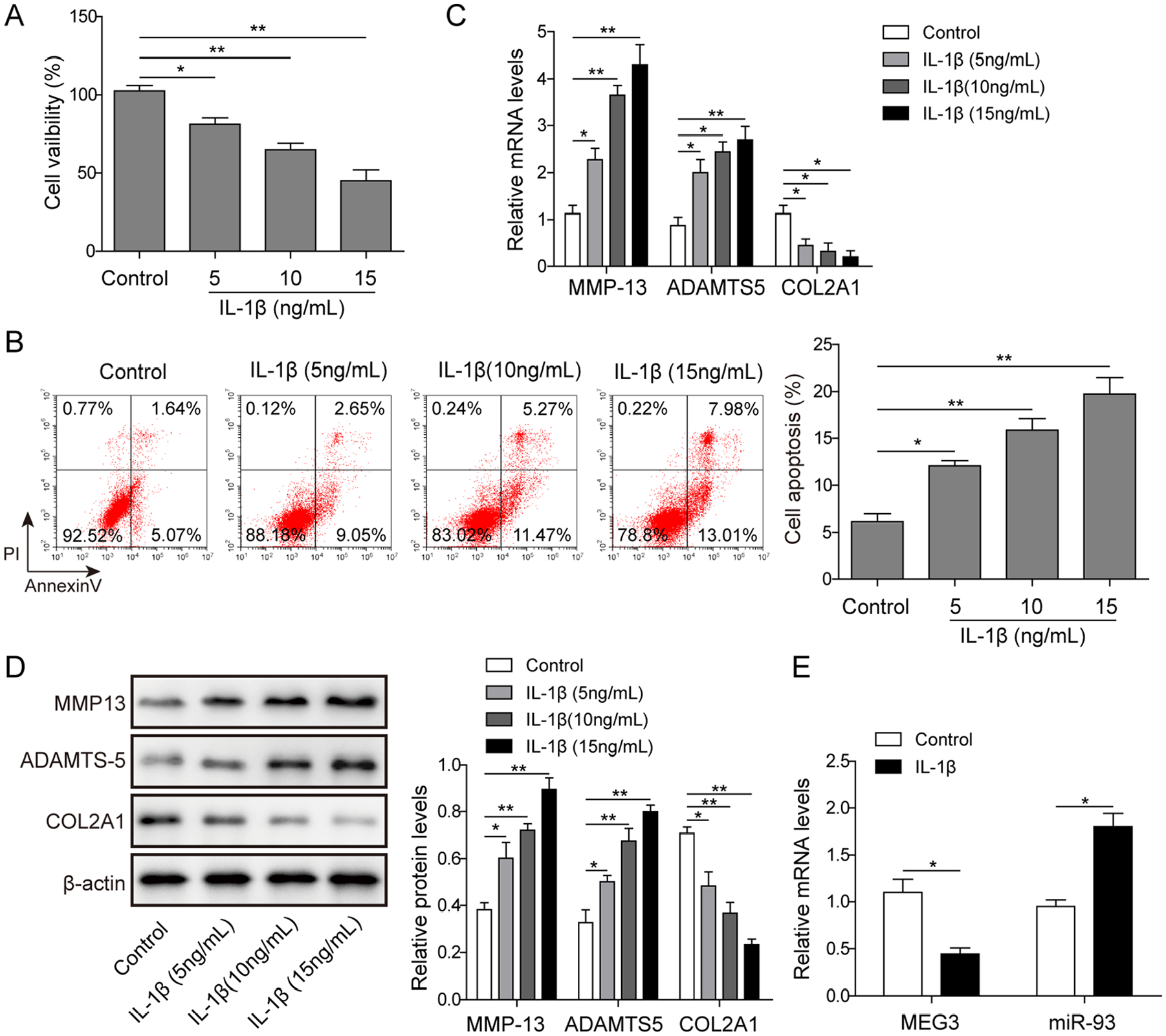
The expression of lncRNA MEG3 and miR-93 in interleukin-1β (IL-1β)-induced chondrocytes. (
Overexpression of MEG3 Inhibited IL-1β-Induced Chondrocytes ECM Degradation
To further investigate the causal relationship between MEG3 and chondrocytes ECM degradation, we assessed the proliferation, apoptosis, and expression of ECM degradation–related biomarkers in IL-1β-induced chondrocytes. First, GFP expression vector was transfected to estimate the transfection efficiency. The transfection efficiency of GFP was assessed to be desirable using fluorescent microscope (
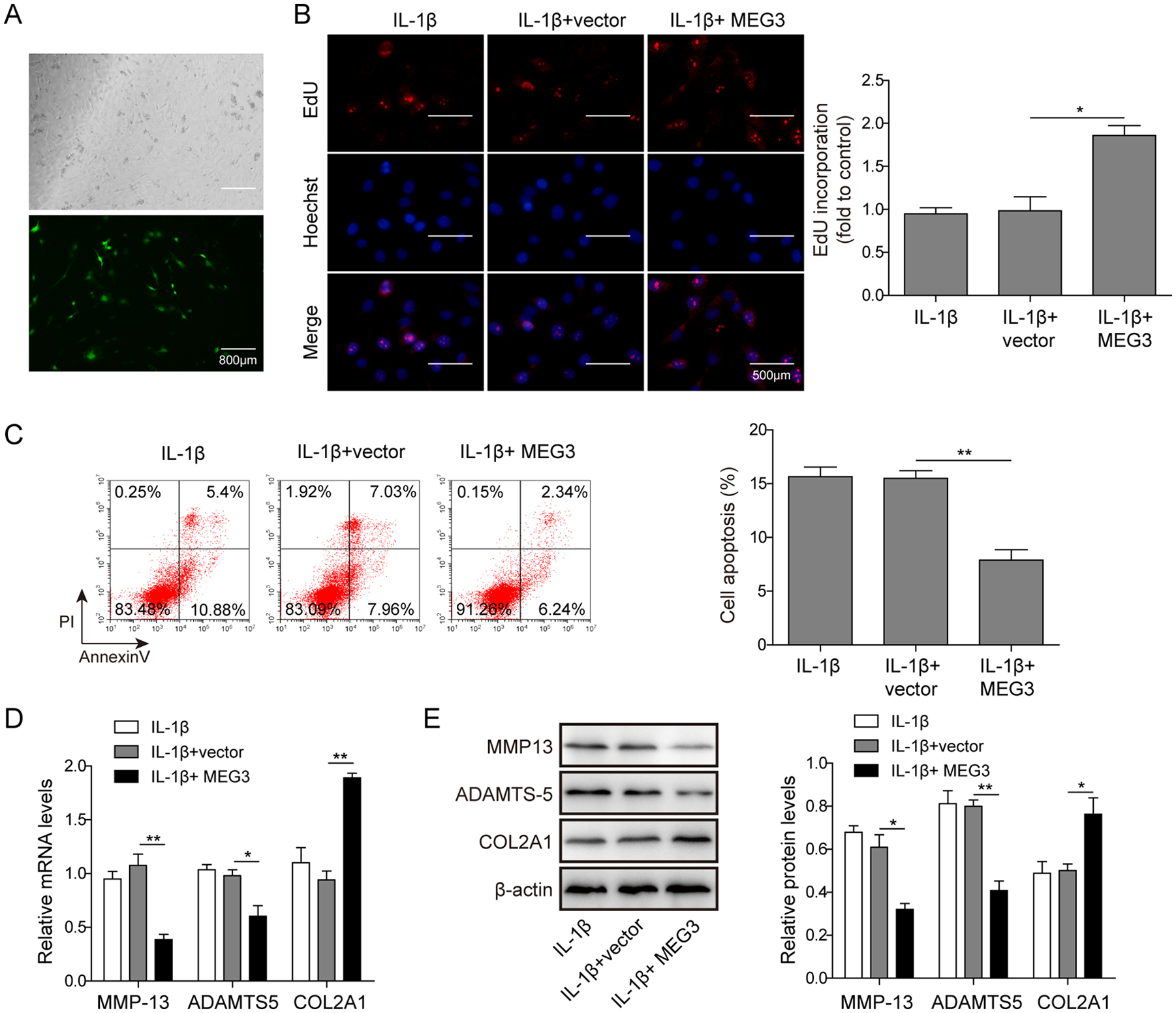
The effect of MEG3 overexpression on the proliferation, apoptosis, and extracellular matrix (ECM) degradation of chondrocytes. (
Knockdown of MEG3 Promoted IL-1β-Induced Chondrocytes ECM Degradation
Base on the above results of gain-of-function study of MEG3, we conducted knockdown experiment of MEG3. EdU assay showed that downregulation of MEG3 suppressed the proliferation of IL-1β-induced chondrocytes by more than 50% (
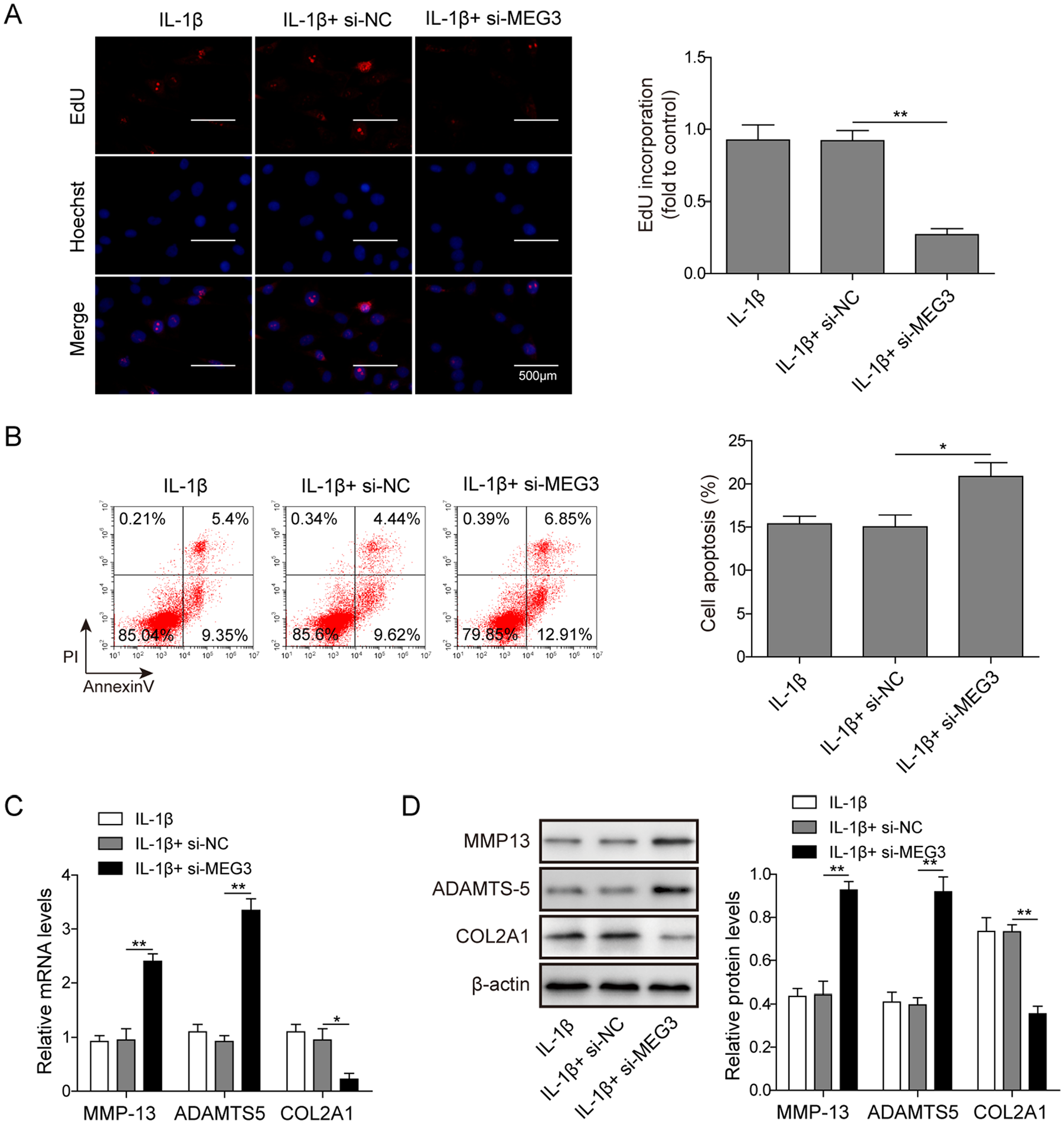
The effect of MEG3 knockdown on the proliferation, apoptosis, and extracellular matrix (ECM) degradation of chondrocytes. (
LncRNA MEG3 and miR-93 Regulated TGFBR2 Expression in Chondrocytes
To further validate the hypothesis that MEG3 targets miR-93/TGFBR2 axis and inhibits the progress of OA subsequently, we explored the regulatory effect of MEG3 and miR-93 on TGFBR2 expression in chondrocytes. Using qRT-PCR, we found that miR-93 expression was suppressed by MEG3 overexpression and induced by MEG3 knockdown (
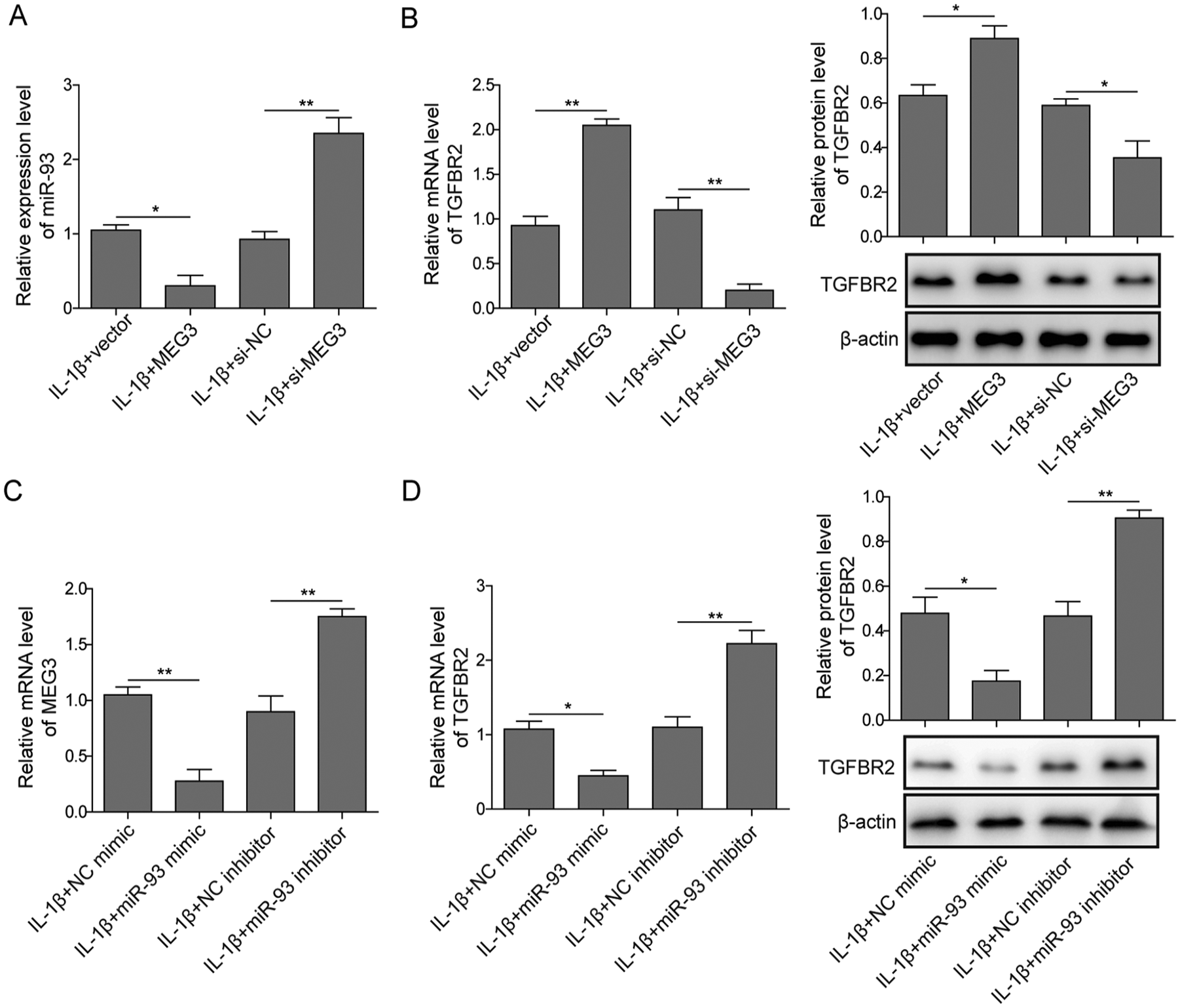
The regulatory effect of MEG3 and miR-93 on transforming growth factor β receptor type II (TGFBR2) expression in interleukin-1β (IL-1β)-induced chondrocytes. (
LncRNA MEG3 Regulated Proliferation, Apoptosis, and ECM Degradation of Chondrocytes via Targeting miR-93/TGFBR2 Axis
To confirm the regulatory effect of MEG3 on ECM degradation in chondrocytes, and the underlying molecular mechanism, we investigated the impact of miR-93 on the protective effect of MEG3 on chondrocytes in OA. We observed that overexpression of MEG3 promoted the proliferation of IL-1β-induced chondrocytes significantly (
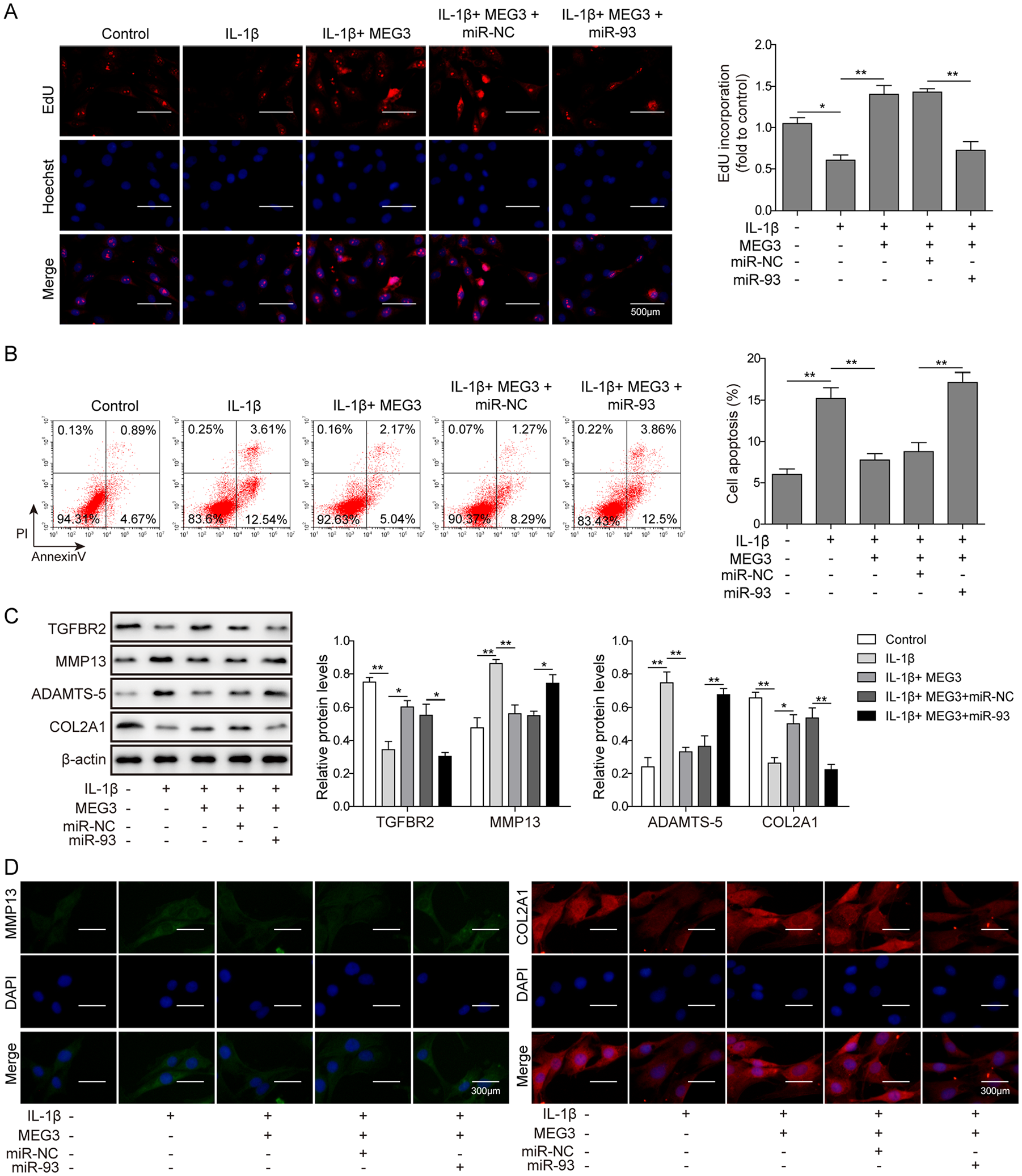
MEG3 regulated extracellular matrix (ECM) degradation in osteoarthritis (OA) via miR-93 and transforming growth factor β receptor type II (TGFBR2). (
Discussion
Articular cartilage is composed of ECM and chondrocytes, which could be stimulated to catabolic and abnormal differentiation by cytokines or growth factors, and further results in ECM degradation subsequently.20-23 The injury or degeneration of cartilage tissue is hard to be self-repaired for its composition characteristics. 24 The process of ECM degradation is complicated and affected by several factors, including genetic, biomechanical, developmental, biochemical, and inflammatory elements. 4 Therefore, the molecular mechanism involved in articular cartilage injury and maintenance need to be further investigated to develop new therapeutic interventions. MMP-13 and ADAMTS-5 play critical roles in ECM degradation in OA as principal enzymes responsible for degradation of aggrecan in articular cartilage. MMP-13 is one of the major enzymes participated in cartilage degradation, targets type II collagen, proteoglycan, type IV and type IX collagen, osteonectin, and perlecan in cartilage. 25 The aggrecanase activity of ADAMTS protein family, especially ADAMTS-5, is involved in pathogenic cartilage degradation. 26 ADAMTS-5 is related with driving cartilage loss and aggrecan degradation in osteoarthritic cartilage.27-30 Moreover, IL-1β also acts as an important inflammatory factor that trigger the apoptosis of chondrocytes, promote the destroy of chondrocytes ECM.31-34 In this study, we treated chondrocytes with IL-1β, which is a common approach to simulate chondrocytes in OA.35-39 Consistently, the chondrocytes treated with IL-1β showed inhibited proliferation, increased apoptosis rate, and differential expression of MMP-13, ADAMTS-5, and COL2A1.
LncRNAs have been demonstrated closely associated with the pathogenesis of OA. Aberrantly expression of lncRNAs have been reported, including HOX transcript antisense RNA (HOTAIR), lncRNA-H19, lncRNA-co-repressor interacting with RBPJ 1 (CIR) and lncRNA MEG3.4,40,41 In addition, MEG3 is closely related with inflammation-related diseases, including OA. It was indicated that MEG3 was involved in OA progression, and the level of MEG3 was inversely associated with vascular endothelial growth factor (VEGF) levels. 11 Under the induction of methylene blue (MB), a long-term inhibitor of peripheral nerve axons, upregulation of MEG3 relieved the OA-associated pain and inflammation by inhibiting P2X3 expression in rabbit. 42 Moreover, MEG3 was found downregulated in the cartilage tissues of OA rat model, and led to the progression of OA via miR-16/SMAD7 axis. 12 In line with the previous findings, the current study showed that MEG3 was remarkably decreased in chondrocytes induced by IL-1β. Besides, MEG3 knockdown significantly inhibited the proliferation, promoted apoptosis, and induced ECM degradation in IL-1β-induced chondrocytes. In addition, overexpression of MEG3 showed positive impact on the proliferation and apoptosis, and further suppressed ECM degradation of IL-1β-induced chondrocytes. The result suggested that MEG3 performed an important protective role in the process of ECM degradation and OA development.
Increasing evidences indicate that TGF-β was included in the development of OA. 43 TGF-β is upregulated in the late phase of OA and stimulates chondrocytes proliferation and proteoglycan synthesis in attempting to repair injured cartilage.44,45 As a transmembrane serine-threonine kinase, TGFBR2 is a major factor to initiate downstream TGF-β signaling. The mRNA level for TGFBR2 was reported dramatically decreased at early stage of OA in a rabbit model. 46 In this study, we proposed to investigate the crosstalk of lncRNA MEG3, miR-93, and TGF-β signaling to provide insights into the molecular mechanism underlying the regulatory function of MEG3 in ECM degradation. We found MEG3 acted as a ceRNA of miR-93, and the overexpression of MEG3 decreased the level of miR-93 by sequence-specific binding. Meanwhile, TGFBR2 can be downregulated by miR-93 as a target. We further determined the regulatory function of MEG3 on TGFBR2, supported the hypothesis that MEG3 increases TGFBR2 by consuming miR-93. Moreover, the overexpression of MEG3 rescued the OA injury simulated by IL-1β in aspects of the proliferation, apoptosis, and ECM degradation in chondrocytes. Thus, the result of current study inferred that MEG3 protected chondrocytes from ECM degradation and OA injury via absorbing miR-93, relieved the inhibited expression of TGFBR2, and then activated TGF-β signaling pathway, suppressed ECM degradation subsequently. Whereas, considering the limitation that IL-1β-induced chondrocytes were used to simulate OA condition in current study, we cannot exclude additional mechanisms such as MEG3 or miR-93 could affect IL-1β or other factors involved in this process directly, although it is less likely to happen.
In summary, based on the OA condition simulated by IL-1β-induced chondrocytes, it was found that lncRNA MEG3 exerted protective function in OA development. Besides, MEG3 regulated TGFBR2 expression indirectly as a ceRNA of miR-93. In addition, MEG3 was able to alleviate ECM degradation and progression of OA through regulating the miR-93/TGFBR2 axis. The conclusion of current study indicated that MEG3 could be a significative biomarker and novel potential target of OA treatment.
Footnotes
Acknowledgments and Funding
We would like to express our sincere gratitude to the reviewers for their constructive comments. The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval for this study was obtained from the Ethics Committee for Animal Research.
Animal Welfare
All rats were treated according to the national guidelines of the care and use of laboratory animals with the approval of the Ethics Committee for Animal Research.