
Abstract
Objective
Osteoarthritis (OA) is an age-related biomechanical and low-grade inflammometabolic disease of the joints and one of the costliest and disabling forms of arthritis. Studies on matrix-degrading enzymes such as metalloproteases, which are implicated in the increased catabolism of extracellular matrix, are of paramount relevance. DKK3 is a member of DKK family and is best known for its role in cancer. Although there is some information about the participation of DKK3 in cartilage pathophysiology and on metalloproteases regulation, in particular, little is known about DKK3 signaling mechanisms. Thus, the aim of this study is to explore how DKK3 regulates matrix metalloproteinase-13 (MMP-13) expression.
Design
Gene, protein expression and protein phosphorylation in primary human chondrocytes and ATDC5 mouse cells were assessed by RT-qPCR and Western blot analysis. Further studies on DKK3 activity were performed by targeting DKK3 gene with a specific siRNA.
Results
DKK3 expression was found to be higher in OA human chondrocytes than healthy cells, being its expression decreased in interleukin-1α (IL-1α)-stimulated cells. DKK3 knockdown increased the induction of MMP-13 elicited by IL-1α in human and mouse chondrocytes and after the analysis of different signalling pathways, we observed that NF-κB pathway was involved in the regulation of MMP-13 expression by DKK3.
Conclusions
Herein we have demonstrated, for the first time, that DKK3 gene silencing exacerbated NF-κB activation, resulting in an increased IL-1α-driven induction of MMP-13. Our results further confirm that DKK3 may play a protective role in OA by attenuating NF-κB activation and the subsequent production of metalloproteases.
Introduction
Osteoarthritis (OA) is the most common form of arthritis and a major cause of musculoskeletal pain and physical disability in the adult population. OA is a degenerative disease that affects the whole joint tissues, including tendons, ligaments, synovium, subchondral bone, and articular cartilage.1,2 The chondrocyte, the unique cell type present in adult articular cartilage, appears as a quiescent cell responsible for the low turnover of the extracellular matrix components under normal conditions. In OA, chondrocytes become “activated,” and are subject to major phenotypical changes and increased production of extracellular matrix-degrading enzymes such as matrix metalloproteinase-13 (MMP-13). 3 This chondrocyte-phenotypic shift is caused, in part, by the exposure to abnormal environmental insults, including altered biomechanical stress and elevated levels of pro-inflammatory cytokines.3,4 One of the most relevant pro-inflammatory cytokines involved in cartilage degeneration is interleukin-1 (IL-1). This cytokine is able to induce in OA chondrocytes a plethora of catabolic mediators, most of them involved in cartilage breakdown, such as MMP-13. IL-1α-driven induction of MMP-13 occurs through the activation of mitogen-activated protein kinases (MAPKs) and different transcription factors such as NF-κB.5,6
Dickkopf WNT signaling pathway inhibitor 3 (DKK3) is a 38-kDa glycoprotein that belongs to the family of DKK proteins. The expression of DKK3 was initially studied in mice and developmental studies revealed a coordinate expression of this protein in several mesenchymal tissues. 7 DKK3 is best known for its role played in cancer. DKK3 is involved in tumorigenesis being downregulated in human immortalized cells and human tumor–derived cell lines. 8 At present, the dysregulation of DKK3 is a consistent and widely shared feature among many cancer types and several lines of evidence describe this gene as a tumor suppressor. 9 However, very little is known about the role of DKK3 in human OA and human cartilage degradation. DKK3 expression was increased in animal experimentally induced OA. 10 DKK3 decreased levels have been found in synovial cells from inflamed zones of synovial membranes compared with normal/reactive areas. 11 Also, it has been recently published that DKK3 can prevent from proteoglycan loss at cartilage level, suggesting a protective role for this protein in OA. 12 This study aimed to test the expression of DKK3 in human and mouse chondrocytes and to study its regulation by a classic pro-inflammatory cytokine such as IL-1α. But the main and most novel goal was to investigate the signaling pathway by which DKK3 modulated cartilage catabolism through the use of knocking-down technique and its effects on MMP-13 expression.
Methods
Reagents
All culture reagents were from Sigma and Lonza. For reverse transcription–polymerase chain reaction (RT-PCR), a First Strand Kit, Master mix, primers for DKK3, MMP-13, and GAPDH were purchased from SABiosciences. Nucleospin kits for RNA isolation were from Macherey-Nagel. Mouse and human recombinant IL-1α and PDTC (pyrrolidine dithiocarbamate) were from Sigma, XAV939 was from Merck. DKK3 siRNAs were purchased from Integrated DNA Technologies.
Cell Culture
Human primary chondrocytes (HAC) and the murine ATDC5 cell line culture were developed as previously described.13,14 Briefly, healthy human articular cartilage samples were obtained from joints of patients with traumatic fractures. Osteoarthritic human cartilage samples were obtained from patients undergoing total joint replacement surgery (with permission from the local ethics committee Galician Ethical Committee, Comité Autonómico de Ética da Investigación de Galicia Secretaria Xeral, Consellería de Sanidade Edificio Administrativo San Lázaro 15703 Santiago de Compostela; COD 2014/310). Informed written consent was obtained from all subjects.
Cartilage samples were obtained from the joint area of the minimal load with a normal morphologic examination (i.e., no change in color and no fibrillation). Human chondrocytes were cultured in Dulbecco’s modified Eagle medium (DMEM)/Ham’s F12 medium supplemented with 10% of fetal bovine serum,
Murine chondrogenic cell line ATDC5, passage 30-50 (purchased from RIKEN Cell Bank), were cultured in DMEM/Ham’s F-12 medium supplemented with 5% fetal bovine serum, 10 µg/mL human transferrin, 3 × 10−8 M sodium selenite, and antibiotics (50 units/mL penicillin and 50 µg/mL streptomycin). ATDC5 cells were differentiated into hypertrophic chondrocytes. Briefly, cells were seeded at a density of 6 × 103/cm2 in 6-well plates with the ATDC5 standard media supplemented with insulin (10 µg/mL). The differentiation media was replaced every 2 days for 14 days. On day 15, the culture medium was switched to α-MEM up to day 21 to obtain hypertrophic cells. Differentiation was qualitatively characterized by increased formation of cell nodules and enhanced staining with Alcian blue, which are indicative of proteoglycan accumulation. In other experiments (data not shown), differentiation was further analyzed by the sequential increase in the levels of type II collagen, aggrecan, and type X collagen mRNA, as previously published. 14
For RT-PCR and Western blot, cells were seeded in P6 multiwell plates until complete adhesion and then incubated overnight in serum-free conditions. Cells were treated with mouse or human IL-1α (0.025, 0.05, or 0.5 ng/mL). NF-κB specific pharmacological inhibitor (PDTC) was added 1 hour before stimulation in a dose of 10 µM.
RNA Isolation and Real-Time Reverse Transcription–Polymerase Chain Reaction (RT-qPCR)
mRNA levels were determined using SYBR-green-based quantitative PCR (qPCR). Briefly, RNA was extracted using a NucleoSpin kit according to the manufacturer’s instructions, and reverse-transcribed (RT) using a SABiosciences First Strand Kit. After the RT reaction, qPCR analysis was performed with a SABiosciences Master Mix and specific PCR primers for: human DKK3 (155 bp, PPH05547F, reference position 1099, GenBank accession no. NM_015881.5); human GAPDH (175 bp, PPH00150E, reference position 1287–1310, GenBank accession no. NM_002046.3); mouse Dkk3 (171 bp, PPM05470A, reference position 1136, GenBank accession no. NM_015814.2); mouse Gapdh (140 bp, PPM02946E, reference position 309, GenBank accession no. NM_008084.2); human MMP-13 (150 bp, PPH00121B, reference position 221-241, GenBank accession no. NM_002427.2); mouse Mmp-13 (88 bp, PPM03675A, reference position 1114, GenBank accession no. NM_008607.1). Amplification efficiencies were calculated for all primers utilizing serial dilutions of the pooled cDNA samples. The data were calculated, using the comparative (ΔΔCt) method and the MxPro software (Stratagene), as the ratio of each gene to the expression of the housekeeping gene. Data are shown as mean ± standard error of the mean (SEM) (error bars) of at least 3 independent experiments and represented as fold-change versus controls. Melting curves were generated to ensure a single gene-specific peak, and no-template controls were included for each run and each set of primers to control for unspecific amplifications.
Western Blot
Whole cell protein extraction was developed using lysis buffer (10 mM Tris/HCl, pH 7.5, 5 mM ethylenediaminetetraacetic acid [EDTA], 150 mM NaCl, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], protease inhibitor cocktail), cell lysates were obtained by centrifugation at 14,000 × g for 20 minutes at 4°C. Nuclear protein extracts were obtained using buffer A (HEPES 10 mM pH 7.9; EDTA 1 mM; ethylene glycol tetraacetic acid [EGTA] 1 mM; KCl 10 mM), ice incubation for 15 minutes, then we added NP-40 0.5% and kept the samples on ice for another 10 minutes. After 15 minutes of centrifugation at 800 × g, we discarded the supernatants and resuspended pellets with buffer C (HEPES 10 mM pH 7.9; EDTA 1 mM; EGTA 1 mM; glycerol 20%; KCl 0.4 M). Samples were then incubated on ice during 30 minutes with occasional vortex. After 15 minutes of centrifugation at 13,000 rpm, we collected the supernatants containing nuclear proteins. Electrophoresis and blotting procedures have been described previously. 15 Immunoblots were incubated with the appropriate antibody (anti-DKK3 diluted 1:1000, Enogene; anti-β-catenin diluted 1:1000, Dako; anti-p65 diluted 1:1000, Santa Cruz; anti-lamin B1 1:1000, Genetex; anti-phospho p38 diluted 1:1000, Millipore; anti-p38 diluted 1:1000, Millipore; anti-phospho Erk 1/2 diluted 1:1000, Cell Signaling Technology, MA, USA; anti-Erk 1/2 diluted 1:1000, Millipore, MA, USA; anti IκB-α diluted 1:1000, Cell Signaling Technology; anti-phospho GSK3β diluted 1:1000, Cell Signaling Technology; anti-GSK3β diluted 1:1000, Cell Signaling Technology) and visualized using an Immobilon Western kit (Millipore) and anti-goat (Santa Cruz) or anti-rabbit (GE Healthcare) horseradish-peroxidase-labeled secondary antibody diluted 1:2000. To confirm equal loading for each sample, after stripping in glycine buffer at pH 3, membranes were reblotted with anti-β-actin antibody diluted 1:5000 (Sigma). Autoradiographs were analyzed with an EC3 imaging system (UVP).
siRNA Transfection
For siRNA-mediated experiments, human primary chondrocytes or ATDC5 cells were seeded at 2 × 105 cells per well in 6-well plates and incubated overnight with DMEM/Ham’s F12 with 10% FBS. The medium was then changed to serum and antibiotics free medium. Transfections were performed following manufacturer’s instructions. Gene silencing was made using 10 nM of mouse DKK3 siRNA or 10 nM of human DKK3 siRNA, we also used a nontargeting control to verify the specificity of the DKK3 gene knockdown. Incubation was continued for 72 hours after siRNA transfection, and the DKK3 knockdown was verified at mRNA and protein levels. At 72 hours after transfection, the cells were treated with IL-1α (0.05 ng/mL). After treatment, total RNA or protein was isolated.
Statistical Analysis
Data are reported as mean ± SEM (error bars) of at least three independent experiments. Statistical analyses were performed by 1-way analysis of variance followed by the Bonferroni test for multiple comparisons, using the GraphPad Prism 4 software, with P values <0.05 considered significant.
Results
DKK3 Is Regulated by OA and Differentiation Status
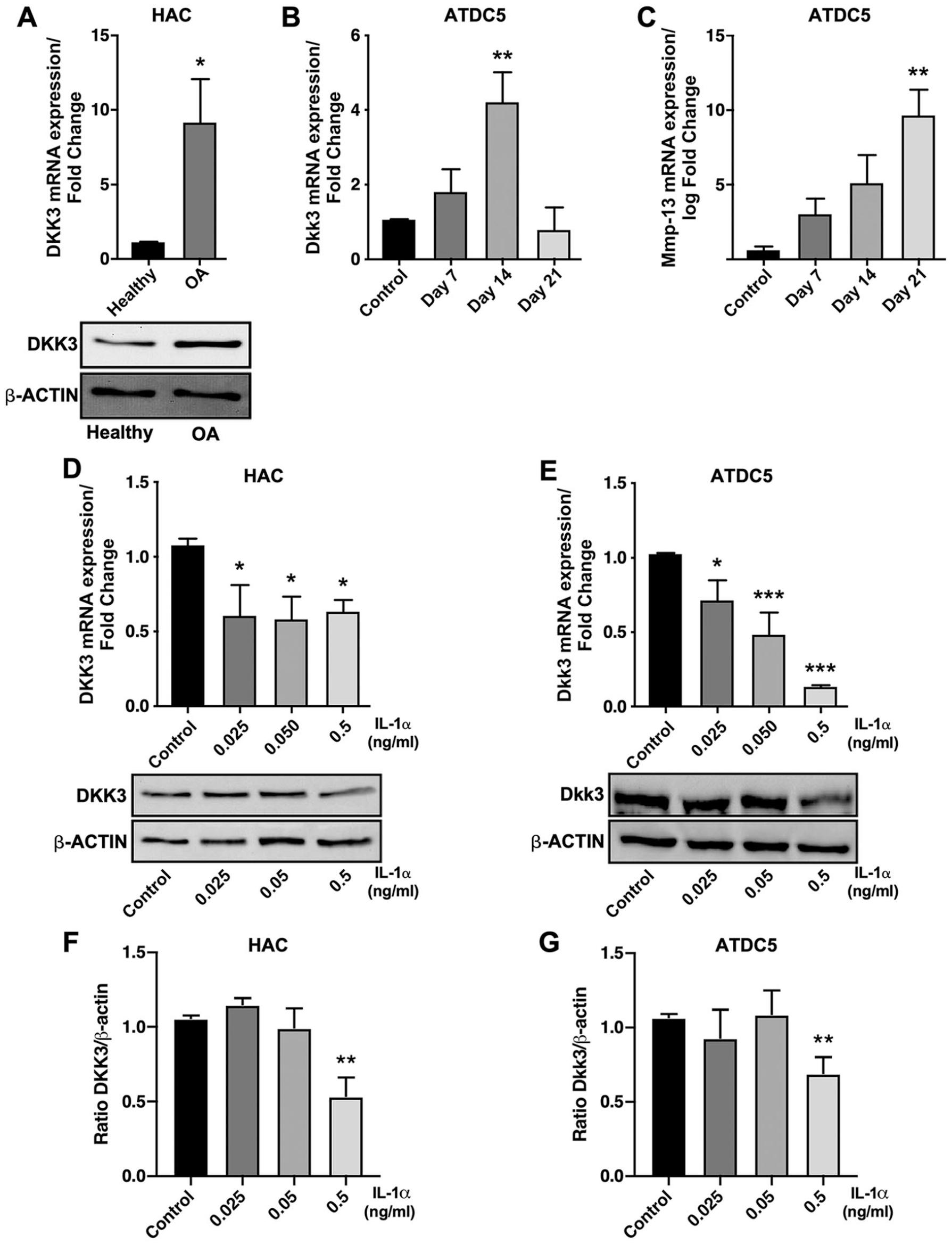
We first wanted to corroborate the observations made by Snelling et al., 12 regarding the expression of DKK3 in OA patients and during chondrocyte differentiation. As shown in Figure 1A , DKK3 expression is increased in chondrocytes from OA patients compared with chondrocytes from healthy individuals. In addition, differentiation of ATDC5 cells, a well-established model of chondrocyte differentiation, revealed that Dkk3 expression had a bell-shaped expression profile being augmented in the first stages of chondrogenesis and reaching a maximum of expression after 14 days of differentiation ( Fig. 1B ). However, Dkk3 expression decreased dramatically at 21 days of differentiation, when cells were hypertrophic ( Fig. 1B ). We observed a progressive increase in the expression levels of Mmp-13, a well-known marker of terminally differentiated hypertrophic chondrocytes, which peaked at day 21 ( Fig. 1C ). Based on that, we showed that Dkk3 expression gradually increased during the chondrocyte maturation process, but this tendency was completely reverted when cells reached hypertrophy. To note, the expression of Mmp-13 during ATDC5 differentiation was found to be the highest when Dkk3 expression declined to minimum levels after 21 days of differentiation ( Fig. 1B and C ).
(
IL-1α Decreases DKK3 Expression
As shown in Figure 1D , all doses of IL-1α exerted a similar effect on DKK3 mRNA expression in human primary chondrocytes. On the contrary, we observed a dose-dependent decrease in the mRNA expression of Dkk3 in ATDC5 cells ( Fig. 1E ), suggesting that undifferentiated chondrocytes were more sensitive to pro-inflammatory stimulation than fully differentiated articular chondrocytes, at least in terms of mRNA regulation. However, analysis of protein expression revealed similar effects in both cell types, only IL-1α at 0.5 ng/mL inhibited DKK3 protein expression ( Fig. 1D and E lower panels, F and G).
DKK3 Regulates IL-1α-Induced MMP-13 Expression
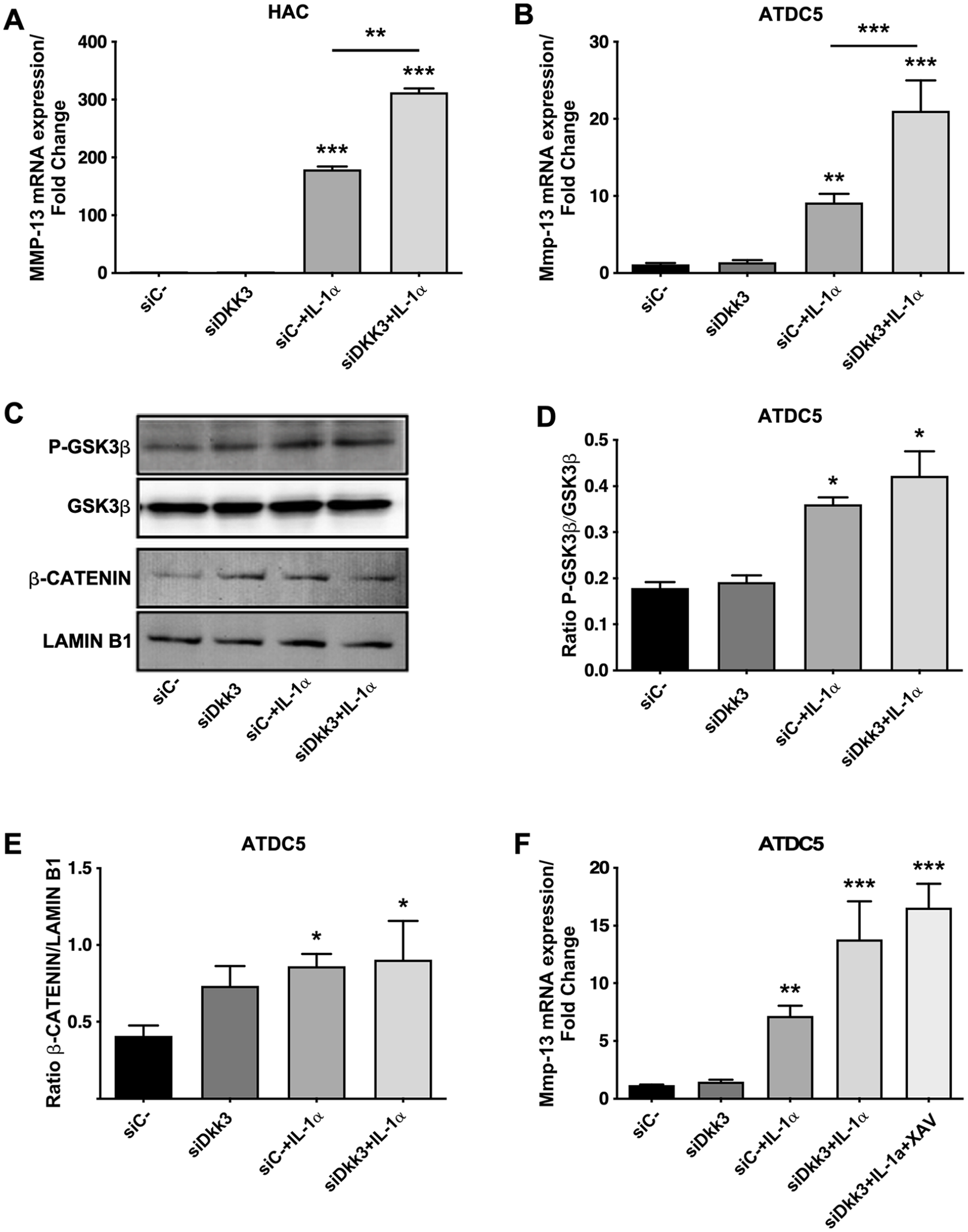
Due to the differential expression of DKK3 in healthy and OA chondrocytes and its regulation by a catabolic factor such as IL-1α, we wanted to ascertain whether DKK3 could have a relevant impact on cartilage catabolism. First, we observed that a siRNA against human DKK3 increased the IL-1α-induced expression of MMP-13 ( Fig. 2A ) in human primary chondrocytes.
(
We also performed Dkk3 silencing in the murine ATDC5 cell line, and similar to that observed for human chondrocytes, we detected that Dkk3 gene knockdown was able to significantly increase the Mmp-13 expression stimulated by IL-1α in ATDC5 cells ( Fig. 2B ).
DKK3 Regulates MMP-13 Expression through NF-κB
Dkk3 is a member of the Dickkopf family proteins, which are well-known Wnt signaling pathway inhibitors. We first tested the ability of Dkk3 to inhibit the canonical Wnt pathway, by analyzing 2 important signal transducers of the canonical Wnt pathway: GSK3β and β-catenin. As shown in Figure 2C and D , Dkk3 gene silencing did not affect GSK3β phosphorylation. As expected, IL-1α clearly induced the phosphorylation of GSK3β. However, Dkk3 gene knockdown in IL-1α-treated cells did not modulate GSK3β phosphorylation ( Fig. 2C and D ). The analysis of β-catenin showed the same pattern to that observed for GSK3β ( Fig. 2C and E ). To confirm that the canonical Wnt pathway was not involved in the observed effects of Dkk3 on Mmp-13 induction by IL-1α, we used the tankyrase inhibitor XAV939, which antagonizes β-catenin signaling. As shown in Figure 2F , the addition of XAV939 did not modulate the expression of Mmp-13, when administered together in Dkk3 knocked-down cells stimulated with IL-1α.
Next, we analyzed whether Dkk3 influenced NF-κB signaling. As shown in Figure 3A and B , Dkk3 knockdown in ATDC5 cells stimulated with IL-1α was able to increase the degradation of IκB-α. Moreover, IL-1α-driven NF-κB subunit p65 translocation to the nucleus was greater in Dkk3 knockdown cells than that observed in nonsilenced chondrocytes ( Fig. 3A and C ). Furthermore, pharmacological inhibition of NF-κB by PDTC abolishes the increase of MMP-13 expression produced by the Dkk3 knockdown in IL-1α-treated cells ( Fig. 3D ).
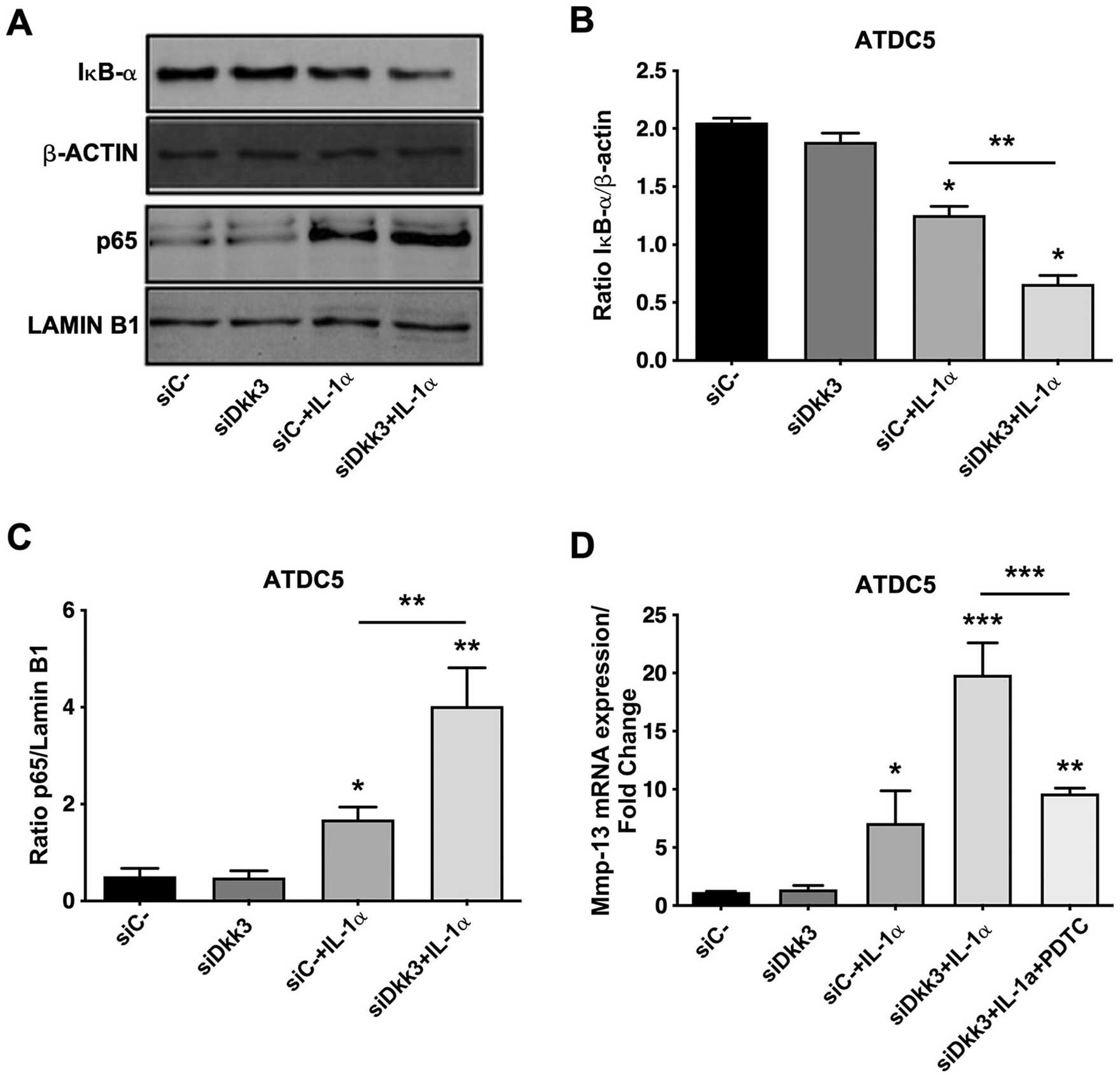
(A) Determination of the protein expression of IκB-α and p65 by Western blot. β-Actin and lamin B1 were used as a loading control. (
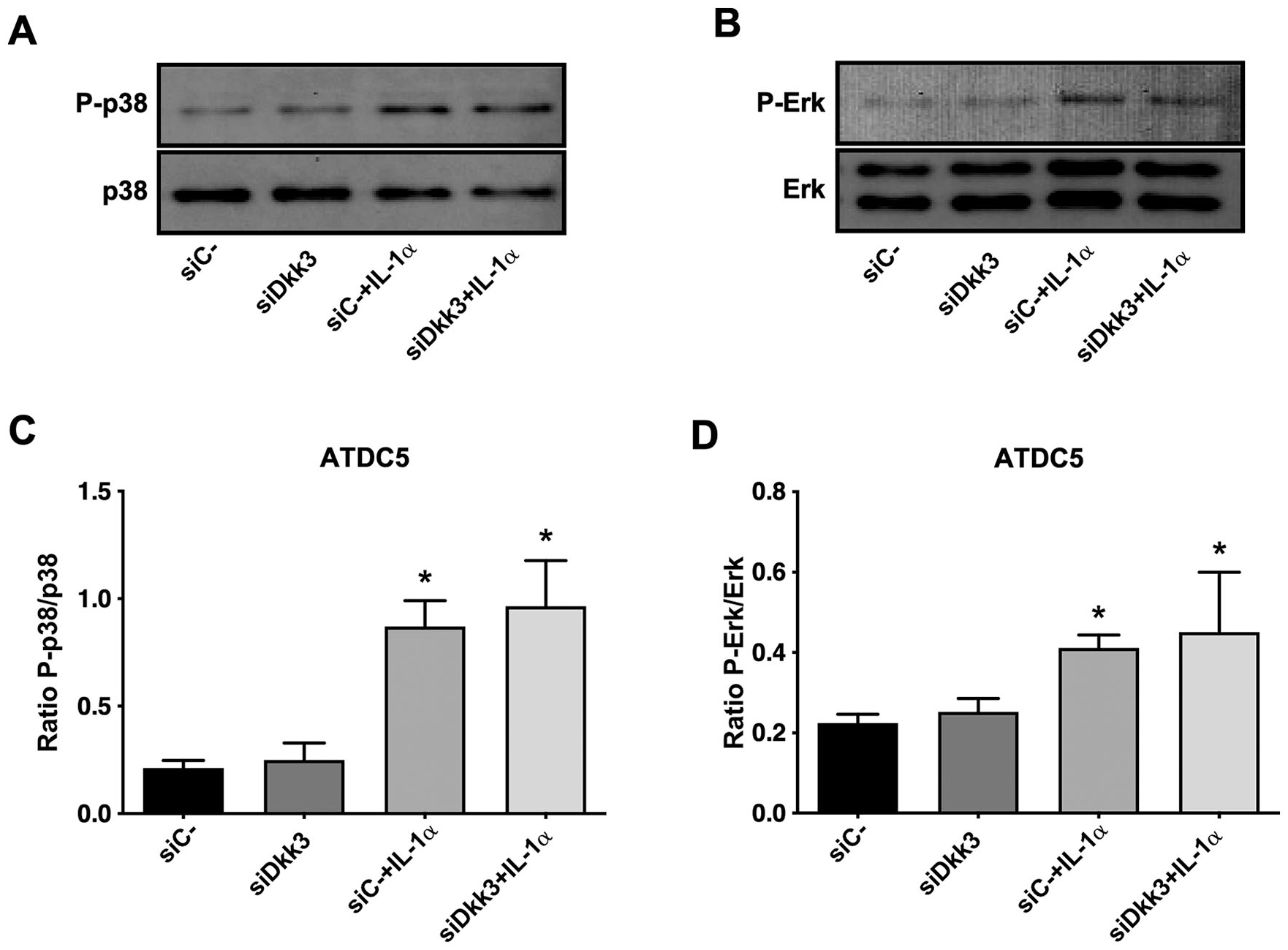
For completeness, we explored whether Dkk3 was able to regulate relevant kinases involved in Mmp-13 induction such as p38 or Erk1/2. As shown in Figure 4A and B , IL-1α treatment phosphorylated both kinases. However, Dkk3 knockdown did not affect the phosphorylation either of p38 or Erk1/2 in IL-1α-treated cells ( Fig. 4A-D ).
(
Discussion
OA is characterized by a progressive loss of articular cartilage. Collagen breakdown by metalloproteinases leads to irreversible degradation of the extracellular matrix, and the participation of MMP-13 as one of the most effective enzymes at degrading type II collagen, highlights a major role for this enzyme in cartilage catabolism.
Recently, the role of Dickkopf family proteins in the development of OA was investigated. DKK1 was found to inhibit cartilage destruction in experimental OA, in part by decreasing MMP-13 production. 16 Regarding DKK3 participation in OA, only a few works showed the involvement of this protein in OA pathology. An upregulation of DKK3 expression in temporomandibular cartilage in experimentally induced OA was reported. 17 Moreover, a study, performed using synovial tissues from OA patients, revealed that synovial cells from inflamed zones showed a downregulation in DKK3 expression in comparison with synovial cells from normal areas. 11 We observed increased DKK3 expression in OA chondrocytes, which is in agreement with a previously published article. 12 Moreover, we also corroborated the observations made by Snelling et al. 12 in which Dkk3 expression increased in the first stages of chondrocyte differentiation and followed by a rapid and dramatic decrease in the hypertrophic stage, matching with a parallel increase in MMP-13 expression. This issue suggested an inverse relationship between these 2 factors that deserved further investigations. For that, we used human primary chondrocytes and the chondrogenic cell line ATDC5, which has been described to be a suitable model for the study of inflammatory-related processes in chondrocytes. This cell line presented more similar behavior to human primary chondrocytes in response to pro-inflammatory stimulus than other chondrogenic cell lines.18,19 First, we observed that IL-1α, a well-known potent inducer of MMP-13, repressed DKK3 expression in chondrocytes, as previously described. 12 Thus, by silencing DKK3 gene, we demonstrated for the first time that DKK3 is acting as a MMP-13 down-modulator in the context of pro-inflammatory cytokine induction, in both human primary chondrocytes and murine ATDC5 cells. This observation is in line with previously published results, which demonstrated that recombinant DKK3 was able to repress the IL-1/oncostatin-M-induced MMP-13 and MMP-1 expression in human primary chondrocytes, and also the proteoglycan release in bovine nasal cartilage explants. 12 Moreover, DKK3 knockdown partially repressed the inhibition of MMP-13 elicited by TGF-β. 12
DKK proteins are Wnt signaling antagonists. For that reason, we examined whether DKK3 modulated MMP-13 expression via Wnt canonical pathway in IL-1α-treated cells. Previously published articles demonstrated how Wnt/β-catenin signaling was involved in the regulation of MMP-13 expression. 20 However, in our study, the analysis of GSK3β and β-catenin revealed that Dkk3 knockdown did not affect the activation of the Wnt canonical pathway elicited by IL-1α. Also, the use of XAV939, as an inhibitor of β-catenin signaling, did not affect the Dkk3 function either. In contrast to other members of Dkk family, whose ability to inhibit canonical Wnt/β-catenin signaling pathway by interacting with Lrp receptors is well known,21,22 the participation of DKK3 in the modulation of this signaling pathway is controversial. Up to now, any receptor for DKK3 has been identified. For that reason, DKK3 ability to influence Wnt signaling routes is thought to be cell and condition dependent. Actually, some effects of this protein might occur through Wnt canonical or noncanonical pathways and even through alternative pathways such as the PI3K/Akt pathway.9,23
In contrast to that observed in human chondrocytes where DKK3 can inhibit Wnt signaling 12 and based on the lack of interaction between Dkk3 and canonical Wnt signaling in ATDC5 cells, we sought to analyze other alternative and novel pathways not yet explored, which might be involved in the regulation of MMP-13 expression. NF-κB is known for its role in the transcriptional regulation of MMP-13 in chondrocytes. 24 As far as we are aware, our study shows, for the first time, a clear relationship between Dkk3 and NF-κB in chondrogenic cells or any other type of chondrocytes. Actually, experimental evidence regarding IκB-α degradation and p65 modulation in Dkk3 silenced cells stimulated with IL-1α is in support of our hypothesis. This aspect may be related to the fact that DKK3 can modify the expression of other transcription factors such as activating transcription factor 6 (ATF6), as in smooth muscle cells. 25
Finally, in order to explore other alternative interactions of Dkk3 with key signal transducers of Mmp-13, we also analyzed, for the first time in chondrogenic cells, the activation of different MAPKs under the stimulation with IL-1α plus Dkk3 knockdown. Other authors observed an interaction between DKK3 and certain kinases such as JNK, Akt or ASK1.23,26 However, in our study, we did not find any variation in the activation of p38 or Erk1/2 after the Dkk3 gene silencing. In agreement with our results, Zhang et al. 26 did not determine differences in the activation of Erk1/2 as a consequence of DKK3 loss- or gain-of-function experiments in cardiomyocytes. In contrast, the same authors have demonstrated an increase in p38 phosphorylation when DKK3 is downregulated. 26 Altogether these data suggest that similar to what happened with Wnt signaling, DKK3 interaction with different kinases might be cell and/or condition dependent.
In conclusion, we demonstrated for the first time the existence of an inverse relationship between the expression of DKK3 and MMP-13 along chondrocyte differentiation. Moreover, DKK3 knockdown resulted in an increase in the induction of MMP-13, one of the most relevant metalloproteases involved in cartilage breakdown, in chondrocytes stimulated with IL-1α. Finally, among the different signaling pathways tested so far, the aforementioned effect likely involves the participation of the NF-κB transcription factor.
Footnotes
Author’s Note
Ali Mobasheri is also affiliated with Research Unit of Medical Imaging, Physics and Technology, Faculty of Medicine, University of Oulu, Oulu, Finland and Departments of Orthopedics, Rheumatology & Clinical Immunology, University Medical Center Utrecht, Utrecht, the Netherlands.
Author Contributions
JC participated in acquisition of data, analysis and interpretation of data and critical revision of the manuscript. CR, VF, RG, AM, MAGG, MS, FL, and JP participated in acquisition of data and samples, drafting of the manuscript and statistical analysis. AM participated in analysis and interpretation of data and drafting/editing the manuscript. OG participated in conception and design of the study, in analysis and interpretation of data, critical revision of the manuscript and scientific supervision of experiments.
Acknowledgments and Funding
OG and FL are Staff Personnel of Xunta de Galicia (Servizo Galego de Saude, SERGAS) through a research-staff stabilization contract (ISCIII/SERGAS). JC is “Miguel Servet” Researcher “CP19/00172 (ISCIII/FEDER), MS and VF are currently “Sara Borrell” Researchers funded by ISCIII and FEDER (CD16/00111). RG is a “Miguel Servet” Researcher funded by Instituto de Salud Carlos III (ISCIII) and FEDER. CR is a predoctoral research scholar funded by ISCIII and FEDER (Exp. 18/00188). OG, RG, and MAGG are members of RETICS Programme, RD16/0012/0014 (RIER: Red de Investigación en Inflamación y Enfermedades Reumáticas) via Instituto de Salud Carlos III (ISCIII) and FEDER. FL is a member of CIBERCV (Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares). The work of OG and JP (PI17/00409), RG (PI16/01870 and CP15/00007) and FL (PI15/00681 PI18/00821 and CB16/11/00226) was funded by Instituto de Salud Carlos III and FEDER. OG is a beneficiary of a project funded by Research Executive Agency of the European Union in the framework of MSCA-RISE Action of the H2020 Programme (Project number 734899). OG is beneficiary of a project funded by Xunta de Galicia, Consellería de emprego e industria (GAIN) (IN607B2019/10). RG is beneficiary of a project funded by Mutua Madrileña 2018. AM wishes to acknowledge financial support from the European Structural and Social Funds through the Research Council of Lithuania (Lietuvos Mokslo Taryba) according to the activity ‘Improvement of researchers’ qualification by implementing world-class R&D projects’ of Measure No. 09.3.3-LMT-K-712 (grant application code: 09.3.3-LMT-K-712-01-0157, agreement No. DOTSUT-215) and the new funding programme: Attracting Foreign Researchers for Research Implementation (2018–2022). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study was approved by the Galician Ethical Committee, Comité Autonómico de Ética da Investigación de Galicia Secretaria Xeral, Consellería de Sanidade Edificio Administrativo San Lázaro 15703 Santiago de Compostela; COD 2014/310).
Informed Consent
Informed written consent was obtained from all subjects.
Trial Registration
Not applicable.