
Abstract
Objective
Epigenetic modifications of DNA are regarded as a crucial factor for understanding the molecular basis of complex phenotypes. This study aims to uncover insight into the epigenetic modifications for Kashin-Beck disease (KBD) by integrating genome-wide association studies (GWAS), methylation quantitative trait loci (meQTLs), and DNA methylation profiles data.
Design
The knee articular cartilages of 5 KBD patients and 5 healthy controls were collected for DNA methylation profiling, using Illumina Infinium HumanMethylation450 BeadChip. Mass spectrograph validation of identified differently methylated genes was conducted using independent samples of 4 KBD patients and 3 healthy controls, together with a previous sample of 2743 Han Chinese individuals of GWAS study for KBD and a study of 697 normal subjects for meQTLs annotation datasets. KBD GWAS single nucleotide polymorphisms (SNPs) and normal meQTLs SNPs were integrated with DNA methylation profiles of KBD articular cartilage to identify genetic control (GC) genes of DNA methylation for KBD. Quantitative polymerase chain reaction (qPCR) was performed to validate the mRNA expression of several identified candidate genes.
Results
A total of 162 CpG sites, 253 SNPs, and 123 GC genes for KBD were identified. Enrichment analysis detected 642 marked GO terms and 19 KEGG pathways (P < 0.05). Six potential key GC genes were conducted for qPCR experiment (ERG, MN1, MITF, WISP1, TRIO, and NOSTRIN).
Conclusions
The results suggest that GC genes of DNA methylation may lead to the erosion of cartilage in KBD, which may help us in understanding the epigenetic alteration of KBD.
Introduction
Kashin-Beck disease (KBD) is an endemic chronic osteochondropathy, which is prevalent at the Eastern Siberia of Russia, North Korea, and the broad diagonal belt from northeast to southwest in China. 1 In 2015, 37.9 million people living at KBD prevalent counties are at the risk of KBD, and 0.57 million patients are suffered KBD in China, including 12,730 children younger than 13 years. KBD is mainly characterized as cartilage degeneration, cartilage matrix degradation, chondrocyte necrosis and apoptosis in growth plate cartilage and articular cartilage. The typical symptoms of KBD include serious joint pain, limited motion, and deformities of joints. According to the diagnosis criteria of KBD in China (WS/T 207-2010), KBD is clinically divided into 3 grades (grades I, II, and III). Recent studies demonstrated the importance of genetic factors in the development of KBD. Nearly half of KBD risk can be explained by genetic components. 2 Several susceptibility genes have been identified for KBD, such as HLA-DRB1, GPX4, and ITPR2.3-5 Additionally, genome-wide RNA expression profiling of KBD articular cartilage observed altered RNA expression profiles and reported a group of differently expressed mRNA, microRNA, and lnRNA molecules for KBD.6-8 However, the molecular mechanism of KBD remains unclear. A large part of grade III KBD patients finally need total knee replacement, resulting in heavy medical and economic burden to the patients.
Epigenetic modifications of DNA are regarded as a crucial factor for understanding the molecular basis of complex phenotypes. 9 DNA methylation is one of the major epigenetic modifications that plays a pivotal role in gene expression regulation. Recent studies have demonstrated the implication of DNA methylation alteration in the development of arthritis. 10 Jeffries et al. 11 performed a genome-wide DNA methylation profiling of cartilage in osteoarthritis (OA) patients and healthy controls, identifying multiple differently methylated genes and correlation between CpG methylation and histologic severity in OA. Another study identified 1,859 differentially methylated loci in the fibroblast-like synoviocytes (FLS) of rheumatoid arthritis (RA), indicating DNA methylation altering the gene expression levels of FLS and leading to the joint damage of RA. 12
Moreover, recent studies observed that DNA methylation status is under genetic control. The regulatory genetic variants affecting DNA methylation levels are named as methylation quantitative trait loci (meQTLs). 13 It has been identified that peripheral blood DNA methylation associated with the risk of some malignancies even causing mortality. 14 Accordingly, a large amount of meQTLs have been detected and highlighted for complex diseases. In addition, it has been found that the disease-associated loci identified by GWAS were enriched in regulatory genetic variants. 15 Therefore, integrative analysis of GWAS, meQTLs, and DNA methylation profiles data may uncover novel insight into the epigenetic modifications for complex diseases. In this study, we conducted a trans-omics integrative analysis of KBD GWAS and KBD DNA methylation profiles to detect novel candidate methylation genetic control (GC) genes and their corresponding pathways for KBD.
Methods
Ethics Approval and Consent of Participants
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Human Ethics Committee of Xi’an Jiaotong University (project identification code: 2015-237). A written informed consent was obtained from all the patients or relatives of donors.
Study Subjects
A total of 9 grade III KBD patients and 8 healthy control subjects, matched for age and sex were enrolled for genome-wide DNA methylation profiling (5 grade III KBD patients and 5 control subjects) and mass spectrograph validation (4 grade III KBD patients and 3 control subjects). All the KBD patients and healthy control subjects were Chinese Han. The clinical data of study subjects were obtained from nurse-administered questionnaires, including self-reported ethnicities, lifestyle characteristics, health statuses, family, and medical histories. KBD was diagnosed according to the diagnosis criteria of KBD in China (WS/T 207-2010). Healthy control cartilage specimens were collected from the subjects undergoing amputation caused by traffic accidents. The subjects with genetic bone and cartilage diseases, RA and other skeletal diseases, or arthritis were excluded. The total of 17 articular cartilage specimens were harvested from the same anatomic areas of femoral condyles of knee joints.
DNA Methylation Profiling of KBD and Healthy Controls
DNA was extracted from the cartilage specimens using QIAamp DNA Mini Kit (QIAGEN, Germany) following the standard protocol. Agarose gel electrophoresis was employed to evaluate the quality of extracted DNA. Illumina Infinium HumanMethylation450 BeadChip was conducted for genome-wide DNA methylation profiling of 5 KBD and 5 healthy controls. A total of 500 ng DNA was used for bisulfite conversion according to the standard protocol for the EZ DNA Methylation Kit (Zymo Research, USA). Following addition of whole-genome amplification, the samples were incubated overnight at 37°C. After hybridization to HumanMethylation450 array, the array was washed and stained following the instructions of Infinium HD Assay Methylation. The raw image intensities were obtained by IScan SQ scanner (iScan System, Illumina, USA).
Differentially methylated CpG sites were identified by empirical Bayes moderated t test implemented in limma package. 16 Adjusted P value was calculated by Benjamini-Hochberg method. Significant CpG sites were defined as (1) adjusted P value (Padj) ≤0.05; (2) |β-value difference (Δβ)| ≥ 0.2. For quality control, the CpG sites with missing values, or detection P values >0.05 in more than 90% of cartilage specimens were excluded.
Mass spectrograph validation was conducted using an independent cartilage sample set of 4 KBD patients and 3 healthy control subjects to assure the reliable outcomes of DNA methylation. EpiTect Bisulfate kit (Qiagen, German) was applied to process extracted DNA specimens. Sequenom MassARRAY platform was used for measuring the methylation level of selected CpG sites following standard protocol. Methprimer was used for polymerase chain reaction (PCR) primers design (http://epidesigner.com). The methylation data were generated by Epityper software version 1.0 (Sequenom, CA).
KBD GWAS Data
The GWAS data of KBD were drawn from our published study. 4 In brief, a GWAS of KBD was conducted. Following the extreme phenotype sampling approach, 90 grade III KBD patients and 1,627 healthy control subjects were recruited for GWAS. Peripheral blood samples (5 mL) were obtained from each participant. E.Z.N.A Blood DNA Midi Kit (Omega Bio-Tek) or a Pure gene DNA isolation kit (Gentra Systems) was used to extract genomic DNA from peripheral blood leukocytes. Affymetrix Genome-Wide Human single nucleotide polymorphisms (SNPs) Array 6.0 conducted for genotyping. Data management and quality control curbed by Affymetrix GeneChip Command Console software. PLINK were used for genome-wide association study. The overage call rate of GWAS samples was 98.92%. In terms of quality control, 106,625 SNPs (call rates of <98.00%), 311,776 SNPs (minor allele frequencies [MAF] < 0.01) and 6,000 SNPs (P < 0.001 from Hardy-Weinberg equilibrium [HWE] testing) were excluded. Significant associations were identified at a traditional genome-wide threshold (P< 5.0 × 10−8). After quality control filtering, a total of 532,894 SNPs were retained for the GWAS. Detailed procedures could be found in our published study. 4
meQTLs Annotation Datasets
meQTLs annotation datasets were drawn from a previous study. 13 Briefly, the peripheral blood specimens of 697 normal subjects were collected. Next-generation sequencing was conducted to assay blood DNA methylation at approximately 4.5 million loci, and methylation measures at each locus were checked with 4.5 million SNPs to exhaustively screen for meQTLs. 13 The meQTLs were divided into 3 groups, including 542,061 “local” meQTLs (SNP ≤ 1 Mb from methylation site, corresponding to 25,844 nearby genes), 2,761 “distant-same chromosome” meQTLs (SNP > 1 Mb from methylation site, corresponding to 1,327 nearby genes) and 286 “cross chromosome” meQTLs (corresponding to 156 nearby genes). 13
mRNA Expression in KBD Chondrocytes
Quantitative real-time polymerase chain reaction (qPCR) was used to validate the functional relevance of identified candidate genes in KBD. Human articular chondrocytes were obtained from another independent samples of 3 grade III KBD patients and 3 normal controls with femoral neck fracture (FNF) for qPCR. The collected cartilages tissues were cut into 5 to 10 mm3 slices and incubated with 0.2% trypsin at 37°C for 10 to 20 minutes. After washing with phosphate buffered saline (PBS), the tissue slices were treated for 10 hours with 0.2% type II collagenase at 37°C in constant temperature shaking table. Then, the cells were isolated and cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM)/F12 supplemented with 10% fetal bovine serum (Sijiqing, Zhejiang Tianhang Biotechnology Company, Hangzhou, China) and 1% penicillin/streptomycin. First-passage chondrocytes of all the samples were used in this experiment.
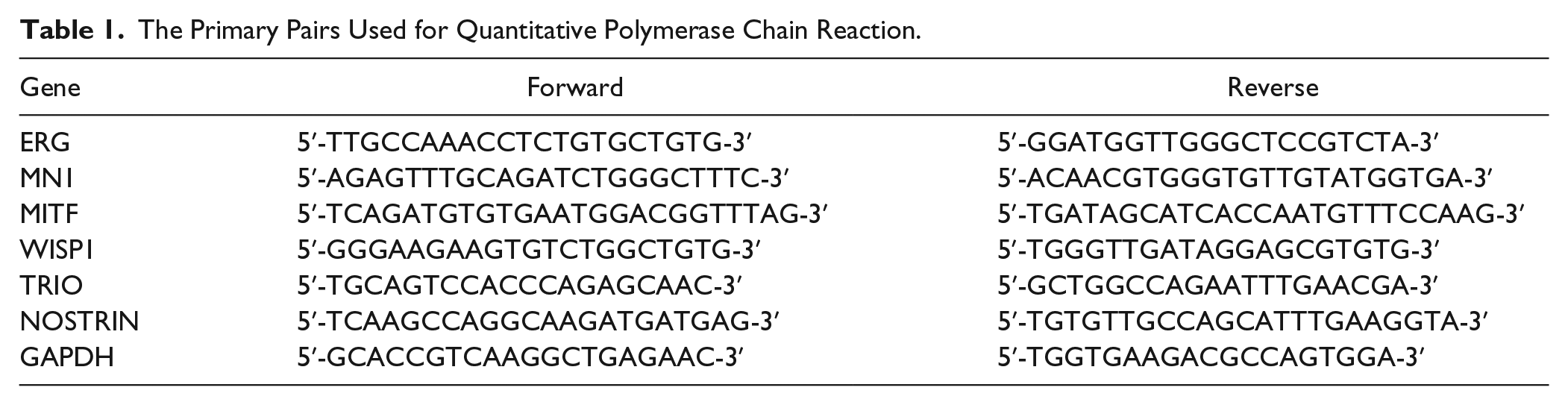
Total RNA from human chondrocytes was isolated according to TRIzol protocol and reverse transcribed to cDNA using One step TaKaRa PrimeScript RT reagent Kit. GAPDH was reference gene. The mRNA expression of genes was detected by qPCR in triplicate analysis, which was performed by specific primers and TaKaRa SYBR Premix Ex Taq II (Tli RNaseH Plus) in the Bio-Rad CFX96 Touch real-time PCR detection system. The reactions were performed in a total volume of 25 µL, containing 2 µL cDNA, 1 µL forward primer (10 µM), 1 µL reverse primer (10 µM), SYBR Premix Ex Taq II (Tli RNaseH Plus) 12.5 µL and sterilized distilled water 8.5 µL. The primer pairs used for qPCR are listed in Table 1 . The cycle of threshold (CT) of each sample was averaged and normalized to that of GAPDH. The results were analyzed by comparative CT equation (2−∆∆CT).
The Primary Pairs Used for Quantitative Polymerase Chain Reaction.
Statistical Analysis
According to SNP ID, the GWAS summary data of KBD was first aligned with the meQTLs annotation datasets to obtain the meQTLs and nearby genes showing association signals (GWAS P < 0.05) for KBD. The identified genes were then compared with the significantly differently methylated genes (detected by DNA methylation profiling) to detect overlapped genes, known as GC genes for KBD. Functional enrichment analysis of the GC genes was performed by ClusterProfiler. 17 T test was performed to compare the mRNA expression levels of 6 candidate genes measured by qPCR.
Results
Genome-wide DNA methylation profiles identified 1,212 different methylated CpG sites and 611 corresponding genes in KBD patients (Padj<0.05). After aligning the GWAS summary data with the meQTLs annotation datasets, we obtained 5,224 “local” meQTLs SNPs (corresponding to 2,059 genes), 12 “distant-same chromosome” meQTL SNPs (corresponding to 4 genes) and 5 “cross chromosome” SNPs (corresponding to 3 genes). Further comparing the differently methylated genes with KBD associated meQTLs SNP lists, we detected 162 CpG sites, 253 SNPs as well as 123 differently methylated GC genes, including 59 hyper-methylated genes and 64 hypomethylated genes in KBD articular cartilage (Supplementary Table S1). All those identified genes are of “local” meQTLs SNPs. Neither “distant-same chromosome” nor “cross chromosome” meQTL SNPs have found corresponding genes related to the DNA methylation of KBD. Six potentially key genes from the identified 123 GC genes were detected by qPCR (ERG, MN1, MITF, WISP1, TRIO, and NOSTRIN). Statistically analysis observed that only the mRNA expressions of MITF (P = 0.0315, ratio = 0.659) and WISP1 (P = 0.0027, ratio = 2.465) are significant in KBD chondrocytes ( Fig. 1 ).
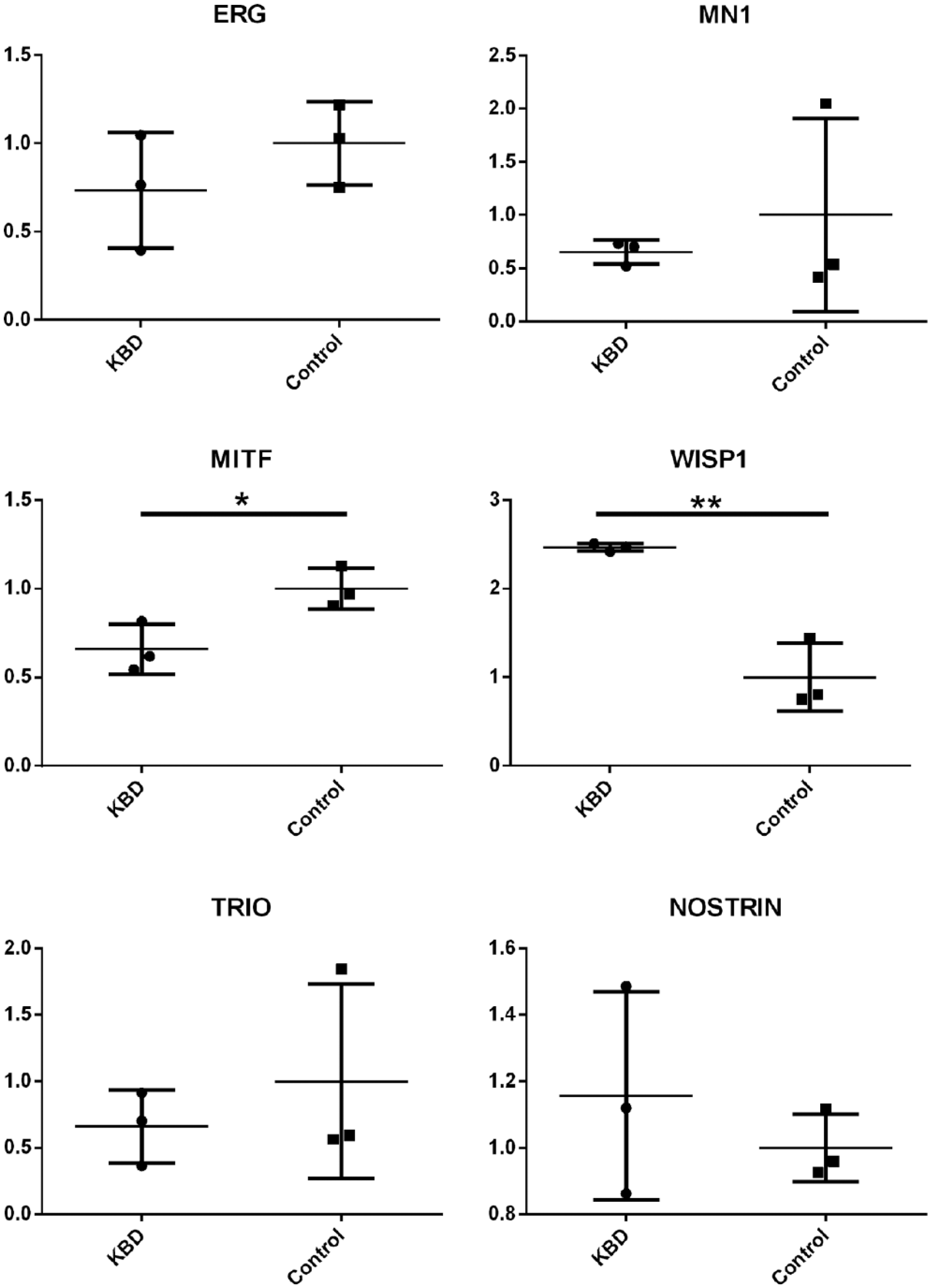
The mRNA expression of ERG, MN1, MITF, NOSTRIN, TRIO, and WISP1, in patients with Kashin-Beck disease (KBD) compared with normal samples.
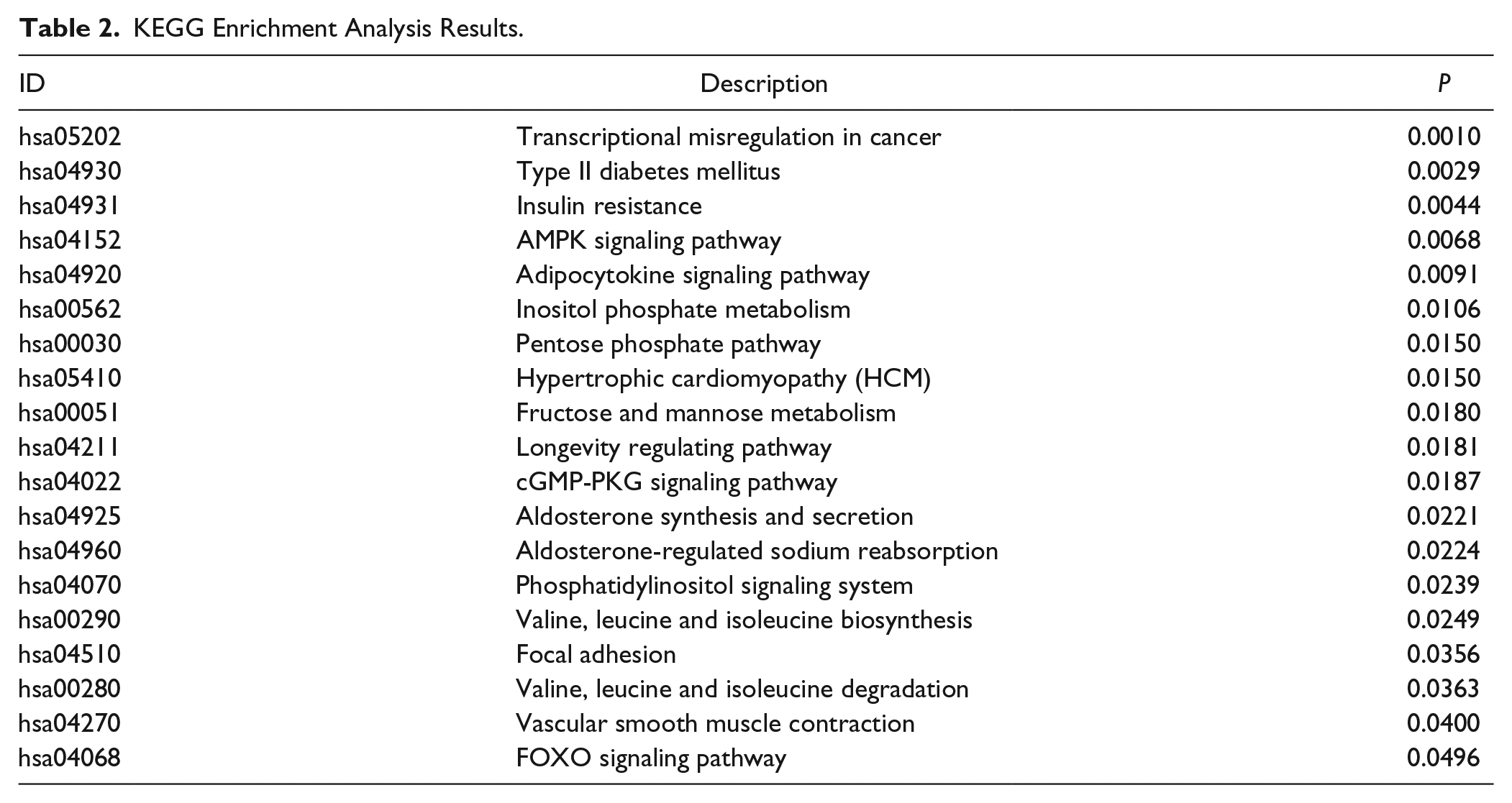
Gene ontology (GO) enrichment analysis detected 624 GO terms (P < 0.05), including 476 GO terms for Biological Process (BP), 37 for Cellular Component (CC), and 111 for Molecular Function (MF) (Supplementary Table S2). The top 10 GO terms are shown in Figure 2 . We observed that neuron migration (GO:00001764, P = 2.20 × 10−5) in BP, cell leading edge (GO:0031252, P = 0.00144) in CC, phosphoric ester hydrolase activity (GO:0042578, P = 0.000361) in MF were the most significant GO terms. Pathway enrichment analysis identified 19 KEGG pathways (P < 0.05), such as AMP-activated protein kinase (AMPK) signaling pathway (hsa04152, P = 0.006791), Adipocytokine signaling pathway (hsa04920, P = 0.0091) and the forkhead box O (FOXO) signaling pathway (hsa04068, P = 0.0496) ( Table 2 ).
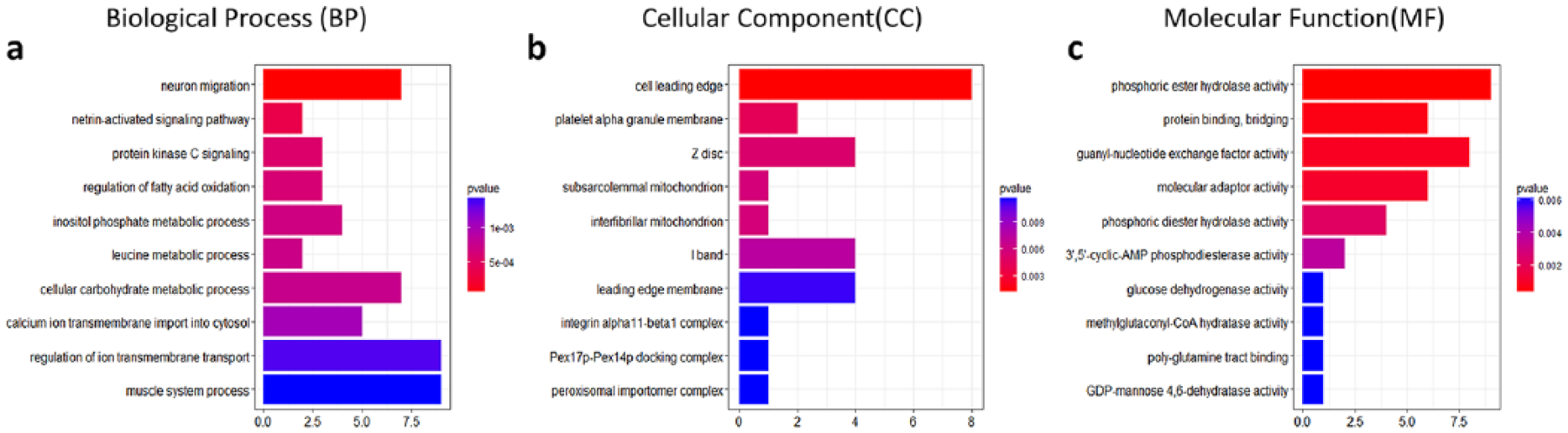
The top 10 gene ontology (GO) terms in Biological Process (BP), Cellular Component (CC), and Molecular Function (MF)
KEGG Enrichment Analysis Results.
Discussion
In this study, a total of 123 GC genes were identified significantly for KBD, and they were only associated with “local” meQTLs SNPs. Showing that GC genes of methylation occur where SNP and CpG site within 1 Mb of each other in the same chromosome in KBD. A few previous studies have identified the correlation between meQTLs and expression QTLs (eQTLs) and found that only 4.8% 18 and 11.8% 19 were significant as both meQTL and eQTL. This could explain what we have found in the mRNA expression of KBD chondrocytes that only 2 out of 6 GC genes significantly expressed.
Among all the identified 123 genes, 48.0% were hypermethylated genes in KBD chondrocytes, in which, MITF was found as the top significant hypermethylated gene, with 3 methylated CpG sites, including cg18503031, cg13523819, and cg13151171. The significant site is at the locus on chromosome 3 and the associated SNP is rs6782870. It also showed significant lower mRNA expression in KBD chondrocytes than controls. MITF encodes melanogenesis associated transcription factor. Numerous studies have identified roles for MITF in oxidative stress response, cell growth, differentiation, miRNA regulation, and DNA methylation.20-22 A model has illustrated that lower MITF levels favor cell invasion and suppress proliferation. 20 Strub et al. 23 recognized that knocking down MITF led to DNA damage, finally ending in cellular senescence. Additonally, Wang et al. 24 have identified oxidative stress mediates the initiation and progression of KBD and leads to necrosis of chondrocytes. Taken together, MITF may play a vital role in the progression of KBD, probably involving in the oxidative stress response, cell growth, and DNA damage in chondrocytes of KBD.
Meningioma 1 (MN1), known as transcriptional regulator, is another significant hypermethylated GC gene for KBD. The significant site is at the locus on chromosome 22 and the associated SNP is rs134133. Its methylated CpG site is cg01214458 in KBD chondrocytes. Prior study has shown that MN1 is a novel target in osteoblastic cells and overexpressed dramatically during differentiation of primary osteoblastic cells, 25 suggesting that MN1 may have pivotal roles in modulating osteoblast proliferation and inhibiting cell growth. KBD is a complicate disorder with skeletal growth and development failure, including osteopenia and osteoporosis. 26 Therefore, more biological research on MN1 might identify the mechanism of osteopenia and osteoporosis in KBD.
WISP1 is a GC gene with hypomethylation. Its methylated CpG site is cg10191240 in KBD chondrocytes. The significant site is at the locus on chromosome 8 with the associated SNP rs4736441. It showed pronounced overexpression in KBD chondrocytes. WISP1 is known as WNT1 inducible signaling pathway protein 1 and the fourth member of the CCN family (cysteine-rich angiogenic protein 61, connective tissue growth factor, nephroblastoma overexpressed). It is considered to have the functions of cell differentiation, cell proliferation, tissue regeneration, and the regulation of transduction.27,28 Wnt signaling pathway plays an important role in embryonic development of cartilage and bone, and the expression of various Wnt ligands is strongly increased in the synovium in experimental OA. 29 It has also found that overexpression of WISP1 induces matrix metalloproteinase and aggrecanase expression and contributes to cartilage damage. 29 In addition, several studies have been identified the abnormal activation of Wnt signaling pathway in KBD. 30 In this study, DNA methylation profiling, GWAS, meQTLs, and mRNA expression have confirmed the hypomethylated and overexpression of WISP1 in KBD patients.
NOSTRIN, a GC gene with hypomethylation, named as nitric oxide synthase (NOS) trafficking, is well known for its relation to nitric oxide (NO). The significant site is at the locus on chromosome 2, with the associated SNP rs17808106, and the methylated CpG sites cg00174992 in KBD chondrocytes. NO regulates a variety of biologic processed such as neurotransmission, inflammatory response, and vascular homeostasis. It stimulates metalloproteinase production and activity, increases susceptibility to cell damage by reactive oxygen species (ROS), inhibits collagen and proteoglycan synthesis, and induces the apoptotic cascade. 31 The enzyme system responsible for producing NO is NOS, which exists in three distinct, such as constitutive neuronal NOS (nNOS), inducible NOS (iNOS) and constitutive endothelial NOS (eNOS). 32 NOSTRIN binds the enzyme responsible for NO production and inhibits production of NO by suppressing the activation of eNOS. 33 eNOS has been identified as acting a mechanistic role in the function of osteocytes, osteoblasts, and osteoclasts as well as chondrocytes. 31 Li et al. 34 found that NO levels significantly increased in adult KBD patients. Together, we speculated NOSTRIN-eNOS-NO may participate in the cartilage damage of KBD.
Pathway enrichment analysis detected AMPK signaling pathway significant in KBD. AMPK is a serine threonine kinase that is highly conserved through evolution and acts as an intracellular sensor modulating the energy balance within the cell. 35 The signaling pathways accelerating AMPK activation have been link to several catabolic responses in diseases. 36 Zhou et al. 37 found AMPK deficiency in chondrocytes aggravated the instability-induced and aging-associated OA in adult mice. Additionally, the AMPK-mTOR signaling pathway is vital in autophagy, which contributed to the imbalance of articular cartilage homeostasis and chondroprotection, leading to cartilage erosion during OA. 38 Therefore, more studies are required to investigate the potential roles and mechanism of AMPK signaling pathway in the cartilage damage of KBD.
It is notable that the FOXO signaling pathway was enriched in KBD patients. The transcription factors of FOXO family regulate the expression of genes in cellular physiological events including apoptosis, cell-cycle control, glucose metabolism, oxidative stress resistance, and longevity. 39 FOXO family contains 4 members in mammals, FOXO1, FOXO3, FOXO4, and FOXO6. Akasaki et al. 40 identified FOXO factors is downregulated in human OA cartilage, contributing to increased apoptotic cell death, intracellular ROS generation as well as decreased antioxidant proteins and autophagy-related proteins in human chondrocytes. Increasing apoptotic chondrocytes, defective autophagy, and ROS have been demonstrated playing important roles in cartilage damage of KBD, which meet the functions of FOXO factors to some extent.
New candidate genes and pathways may provide new clues for understanding the mechanisms of KBD. However, this study only tested 6 of the identified GC genes in KBD chondrocytes. Therefore, it is worthy of more studies to illustrate all those GC genes and associated pathways in the evolution of KBD in future work.
Conclusions
Integrating GWAS data of KBD, meQTL of normal subjects and DNA methylation profiles of KBD, we have identified 123 GC genes significantly for KBD. Our results suggest that GC genes of DNA methylation may lead to the cartilage erosion in KBD. In order to interpret associations with genetic variants or disease phenotype, 123 GC genes may suggest mechanisms in the alteration of DNA methylation in KBD and may help us in understanding the epigenetic alteration of KBD.
Supplemental Material
Additional_Table_S1_5 – Supplemental material for Integrative Analysis of Genome-Wide Association Studies and DNA Methylation Profile Identified Genetic Control Genes of DNA Methylation for Kashin-Beck Disease
Supplemental material, Additional_Table_S1_5 for Integrative Analysis of Genome-Wide Association Studies and DNA Methylation Profile Identified Genetic Control Genes of DNA Methylation for Kashin-Beck Disease by Ping Li, Cuiyan Wu, Xiong Guo, Yan Wen, Li Liu, Xiao Liang, Yanan Du, Lu Zhang, Mei Ma, Shiqiang Cheng, Bolun Cheng, Sen Wang and Feng Zhang in CARTILAGE
Supplemental Material
Additional_Table_S2_GO_Enrichment_P_value_1 – Supplemental material for Integrative Analysis of Genome-Wide Association Studies and DNA Methylation Profile Identified Genetic Control Genes of DNA Methylation for Kashin-Beck Disease
Supplemental material, Additional_Table_S2_GO_Enrichment_P_value_1 for Integrative Analysis of Genome-Wide Association Studies and DNA Methylation Profile Identified Genetic Control Genes of DNA Methylation for Kashin-Beck Disease by Ping Li, Cuiyan Wu, Xiong Guo, Yan Wen, Li Liu, Xiao Liang, Yanan Du, Lu Zhang, Mei Ma, Shiqiang Cheng, Bolun Cheng, Sen Wang and Feng Zhang in CARTILAGE
Footnotes
Author Contributions
FZ takes responsibility for the integrity of the work as a whole, from inception to the finished manuscript. GX gave the conception, design and some critical suggestion to this study. PL, CYW and YW performed all the experiments and analysis. LL, XL, YND, LZ, MM, SQC, BLC, and SW partially performed some experiments. PL and CYW convinced the project and wrote the manuscript.
Acknowledgments and Funding
The work was supported by National Natural Science Foundation of China Grant Numbers 81472925, 81673112, and 81502766; Key Projects of International Cooperation among Governments in Scientific and Technological Innovation Grant Number 2016YFE0119100; and the Fundamental Research Funds for the Central Universities. We would like to thank the authors who generously shared their data as well as all study participants.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Human Ethics Committee of Xi’an Jiaotong University (project identification code: 2015-237).
Informed Consent
A written informed consent was obtained from all the patients or relatives of donors.
Trial Registration
Not applicable.
Supplemental Material
Supplementary material for this article are available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.