
Abstract
Objective:
To evaluate the effects of adipokines and insulin on intracellular calcium concentration ([Ca2+]i) and pH (pHi) in human articular chondrocytes from healthy (CHC) and osteoarthritic cartilage (COC).
Design:
pHi and [Ca2+]i were measured using BCECF and Fura-2 fluorometric probes in CHC and COC under control conditions and following a hypotonic shock. The effects of interleukin-1β (IL1β), tumor necrosis factor-α (TNFα), insulin, leptin, resistin, and adiponectin were assessed.
Results:
pHi was lower in COC than in CHC. Only IL1β β decreased pHi in both cell types; all the agents enhanced pHi recovery following an ammonium prepulse in CHC, effect that was attenuated by Na+–H+ exchanger inhibitors, but they had no effect in COC. Hypotonic shock (HTS) caused a pHi increase, which was significantly smaller in COC. All the hormones attenuated this response and the effect of IL1β was greater. The basal [Ca2+]i was similar in COC and CHC; IL1β, TNFα, and insulin increased the [Ca2+]i, but leptin, resistin, and adiponectin did not. These effects were greater in COC. This [Ca2+]i increase was dependent on extracellular Ca2+ and attenuated by Na+–Ca2+ exchanger inhibitors. HTS caused a [Ca2+]i increase, which was inhibited by transient receptor potential vanilloid blockers and attenuated by all the hormones tested with the exception of adiponectin.
Conclusions:
These findings may help explain the association between obesity and osteoarthritis, in which these hormones are altered. The responses of CHC and COC are different, which suggests that a modification of pH and Ca2+ homeostasis is part of the osteoarthritis pathophysiology.
Introduction
Osteoarthritis (OA) is a disabling disease that develops because of a misbalance in the synthesis and breakdown of the cartilage matrix, which is performed by articular chondrocytes1,2 and is a key factor in maintaining the characteristics of cartilage, in response to a mechanical load, thus leading to a dysfunctional tissue, inflammation, pain, and the functional impairment of joints.3,4 OA has a growing prevalence all over the world and represents a burden on health services.5,6
Osteoarthritis is associated with obesity,7,8 and this condition worsens the clinical course of OA.5,9 Furthermore, several adipokines have been related to OA. Obesity is associated with an increase in leptin and resistin levels, a decrease in adiponectin levels and an increase in pro-inflammatory cytokines, such as interleukin-1β (IL1β) and tumor necrosis factor-α (TNFα).10,11 The interplay of these hormones is an important issue in understanding the pathophysiological process of obesity and metabolic syndrome12,13 and all the other diseases associated with these conditions, such as OA. 14 For instance, pro-inflammatory cytokines have been clearly associated with OA pathophysiology because these cytokines activate matrix metalloproteases (MMPs), induce the production of free oxygen species and prostaglandins and reduce the expression of collagen type II and aggrecan genes. 15 Specifically, IL1β has been found in osteoarthritic synovial fluid and IL1α may produce impairments in the homeostasis of intracellular calcium and pH in chondrocytes.16,17
Moreover, insulin increases collagen type II synthesis in human healthy and osteoarthritic chondrocytes, 18 but does not change aggrecan expression; insulin also enhances GLUT1 expression, particularly in normal chondrocytes. 19 In cartilage explants, insulin stimulates proteoglycan synthesis; inhibits aggrecanase, prostaglandin and nitric oxide production; and overcomes the inflammatory effects of IL1β.
Adipokines also play a role in the pathophysiology of OA.20,21 A number of studies have found a relationship between increased plasma and articular levels of some adipokines and OA. 22 The cause of the increase of these adipokines in synovial fluid is not clear, although the increase could be attributable to various factors, such as greater synovial membrane permeability, the production of inflammatory agents by synoviocytes and the synthesis of adipokines by the infrapatellar fat pad, in the case of knee OA.
Leptin can be produced by chondrocytes and synoviocytes and can also act in these same cells.23,24 At low doses, leptin induces the growth and proliferation of chondrocytes and extracellular matrix synthesis. At high doses, such as those found in obesity, leptin is proinflammatory, activates MMPs, induces the production of free oxygen species and alters chondrocyte metabolism, thereby inducing extracellular matrix catabolism. 25 Adiponectin also promotes matrix catabolism and has been correlated with the severity of knee OA and aggrecan degradation. 26 Intriguingly, in almost all tissues, adiponectin is anti-inflammatory and protective, even in cartilage 27 ; there are some studies describing an inhibitory effect of adiponectin on MMPs and pro-inflammatory cytokines. Resistin has inflammatory effects in cartilage; induces the expression of other pro-inflammatory cytokines, MMPs, and free oxygen radicals; and promotes matrix catabolism. 28 This hormone has also been associated with a more severe OA. IL1β, a pro-inflammatory cytokine that is also released from adipocytes in obesity and is increased in the synovial fluid of osteoarthritic joints, produces an increase in intracellular calcium in chondrocytes, alters intracellular pH in these cells and may modify chondrocyte homeostasis in osteoarthritic cartilage. 29
However, the effects of other adipokines on the ionic homeostasis of chondrocytes have not been explored although intracellular composition may regulate the extracellular matrix metabolism. 30 This study aims to evaluate the effects of a number of adipokines on [Ca2+]i and pHi, and the pathways involved in those effects, in articular chondrocytes because these specific factors may regulate matrix turnover and are determining in the chondrocyte homeostasis.30-32 The effects of these agents on the response to a hypo-osmotic challenge were also evaluated, given that chondrocytes are subjected to fluctuations in extracellular osmolarity during joint movement, which result from the fluid flux following changes in mechanical load. 33 Furthermore, changes in [Ca2+]i and pHi can be produced in response to these osmotic challenges, as shown in bovine34-36 and equine 37 chondrocytes.
Method
Cartilage and Isolation of Chondrocytes
This study was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans and was approved by the Bioethics Committee of the Universidad Tecnológica de Pereira. Cells were isolated from knee or hip load-bearing cartilage obtained from patients who were undergoing joint replacement surgery because of OA and who signed an informed consent form, regardless of gender or age. The diagnosis of severe OA was made by the orthopedic surgeon via arthroscopy and was confirmed by the joint’s macroscopic appearance during surgery; only the severely affected areas were sampled. The healthy cartilage was obtained from patients without macroscopic signs of degeneration who were subjected to surgery because of trauma, and the cartilage was extracted without any damage to the donor.
Cells were isolated using a method modified from the optimized low-temperature protocol described by Hidvegi et al. 38 Briefly, the cells were isolated by trypsin predigestion (0.5% in HEPES-buffered saline [HBS] for 15 minutes at 37°C) followed by digestion with a mixture of type I and type II collagenase (2000 U/mL each in HBS for 20 hours at 27°C); after the filtration of the isolation solution, the cells were resuspended in fresh 300 mOsm/L Dulbecco’s modified Eagle’s medium (DMEM) prior to resuspension in the experimental media. Previous studies have demonstrated that there are no functional alterations in human and bovine chondrocytes following the digestion protocol.17,35,38-41 All the experiments were performed on freshly isolated cells.
Media and Chemicals
All the chemicals and solutions were obtained from Sigma-Aldrich (St Louis, MO), unless otherwise stated. The cartilage slices and isolated chondrocytes were incubated in DMEM and supplemented with glutamine (2 mmol/L) and antibiotics (100 µmol/L penicillin and 100 µmol/L streptomycin; Life Technologies, Grand Island, NY).
The HBS solution contained (in mmol/L) 145 NaCl, 5 KCl, 2 CaCl2, 15 HEPES, and 10 glucose with the pH adjusted to 7.4 at 25°C using 1 M NaOH. The Ca2+-free solution lacked CaCl2, and EGTA (1 mmol/L) was added. For the Na+-free solution, NaCl was replaced with NMDG-Cl at the same concentration. Hypotonic shock (HTS), 290 to 145 mOsm/L, was induced by a 50% dilution of the solution with distilled water, which was abruptly added to the cell suspension.
Stock solutions of insulin (172 mmol/L), TNFα (2 µg/mL), IL1β (0.1 mg/mL), and adiponectin (0.5 mg/mL) were dissolved in phosphate-buffered saline solution (PBS). Resistin (0.5 mg/mL) was dissolved in a 25 mM Tris and 25 mM NaCl solution with a pH of 7.5, and leptin (1 mg/mL) was dissolved in a 15 mmol/L HCl and 7.5 mmol/L NaOH solution with a pH of 8.0. All the solutions were aliquoted and stored at −20°C.
Measurement of Intracellular pH
The pH was measured according to a method previously described using the pH-sensitive fluorescent dye BCECF. 42 Briefly, the isolated chondrocytes were preacidified with an ammonium prepulse (20 mmol/L NH4Cl in HBS, 15 minutes), to evaluate the pHi recovery, as described by Tattersall et al., 17 the cells were then suspended in HBS and loaded by incubation with BCECF-AM (10 µmol/L for 20 minutes at 37°C) before the fluorescence emission was recorded for 300 seconds at 37°C with magnetic stirring (FP-6500 spectrophotometer, Jasco, Tokyo, Japan). The dye was alternately excited at 490 and 439 nm, and the fluorescence emission was measured at 535 nm. The 490/439 nm signal ratio was calibrated previously using the nigericin/high K+ method described by Thomas et al., 43 before each experiment with a new set of samples. The pHi recovery was measured in cells that had been pre-incubated for 1 hour with all the hormones tested.
The acid equivalent fluxes (JH, mM/min) were calculated for the first 300 seconds of recovery using the following equation:
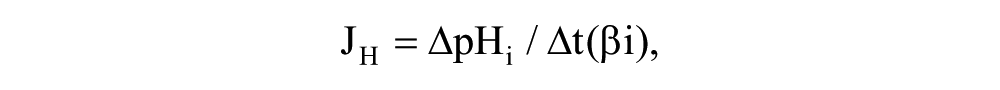
where βi corresponds to the buffering power, which is −43.3pHi + 377 for pHi values between 6.4 and 7.2 for this type of cell as described by Browning et al. 44
To test the effects of the main inhibitors of the pathways involved in H+ transport, the cells were resuspended in a Na+-free solution or in HBS supplemented with Na+–H+ exchanger (NHE) inhibitors, HOE694 (10 μmol/L, specific for NHE1) or amiloride (1 mmol/L, a nonspecific dose) or with the H+-ATPase inhibitors NBD-Cl (chloro-7-nitrobenz-2-oxa-1,3-diazole; 100 µmol/L) or bafilomycin A1 (200 nmol/L). In all cases, chondrocytes were loaded with BCEC-F before being exposed to drugs.
Measurement of Intracellular Calcium Concentration ([Ca2+]i)
The previously isolated cells were loaded with Fura-2 (5 µmol/L) by incubation in HBS for 30 minutes at 20°C followed by 15 minutes at 37°C. 45 The cell suspension was then centrifuged, and the cells were resuspended in the appropriate experimental medium before being transferred to a cuvette. Fluorometer measurements were recorded for 300 seconds at 37°C with magnetic stirring (FP-6500 spectrophotometer, Jasco, Tokyo, Japan). The dye was alternately excited at 380 and 340 nm, and the fluorescence emission was measured at 510 nm. The 380/340 nm signal ratio was calibrated before every experiment using the Grynkiewicz et al. method. 46 Briefly, the fluorescence ratio was measured in HBS lacking CaCl2 and supplemented with EGTA (1 mmol/L) and also in a 2 mmol/L Ca2+ solution of HBS supplemented with ionomycin (300 nmol/L), a concentration of Ca2+ at which Fura-2 is saturated. Maximal and minimal ratios (Rmax and Rmin) were obtained under these 2 conditions, and the [Ca2+]i values were derived using the following equation:
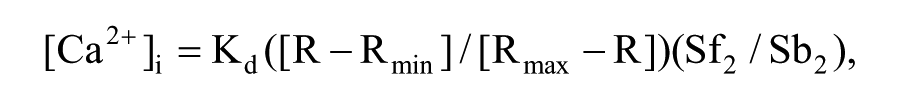
where Kd is the dissociation constant for Fura-2 (224 nmol/L), R is the experimentally measured ratio, Sf2 is the fluorescence measured at 380 nm under the Ca2+-free condition, and Sb2 is the fluorescence measured at 380 nm with saturating Ca2+ (2 mmol/L).
The [Ca2+]i was measured in cells preincubated for 1 hour with all the hormones tested. In all cases chondrocytes were loaded with Fura-2 before being exposed to these agents.
Statistics
The results are presented as the mean ± standard error of the mean, where n is the number of isolation batches. Each isolation batch represents the cells from the cartilage obtained from one patient’s joint. Statistically significant differences were determined using a 2-way analysis of variance test. The Bonferroni method was employed as a post hoc test.
Results
Effects on pHi in CHC
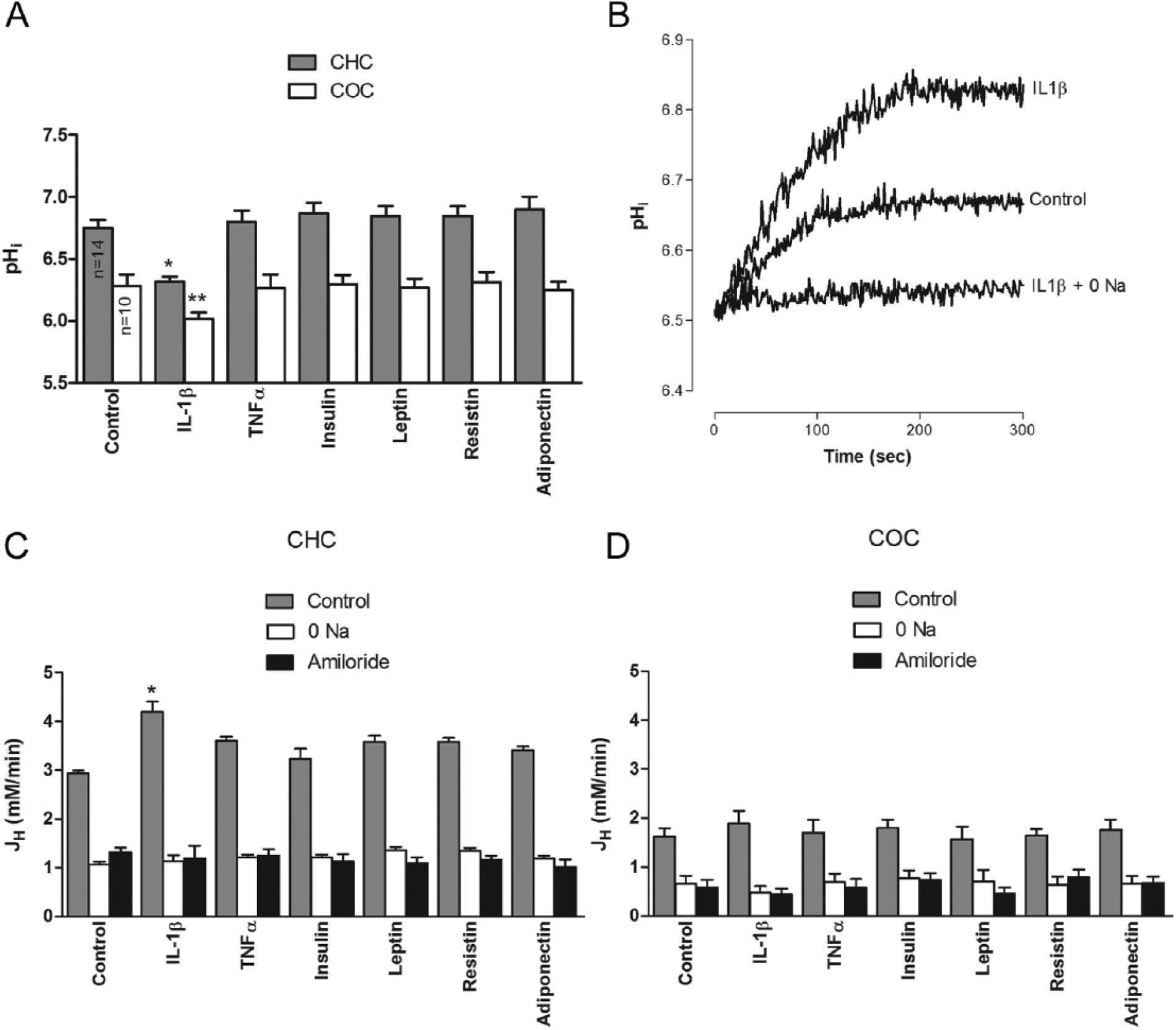
The effects of a number of adipokines and insulin on pHi were recorded over a 300-second period in basal conditions and after an acidifying ammonium prepulse in BCECF-loaded human chondrocytes from healthy cartilage (CHC) treated with IL1β (10 ng/mL), TNFα (10 ng/mL), insulin (100 µmol/L), leptin (100 ng/mL), resistin (10 ng/mL), and adiponectin (100 ng/mL). The doses were those in the range used in several previous studies that explored the cellular effects of these agents.47-51 In all cases, BCECF loading was performed prior to the incubation with these agents. The basal pHi was 6.74 ± 0.21 (n = 18). IL1β, but not TNFα, insulin, leptin, resisti,n or adiponectin, caused a significant decrease in pHi before the ammonium prepulse ( Fig. 1A ), but all of the factors caused a significant increase in the pHi recovery after the ammonium prepulse compared to the control ( Fig. 1B ); this effect was inhibited by the NHE inhibitor amiloride (1 mmol/L) and was dependent on the presence of extracellular Na+ ( Fig. 1C ). However, the effect on pHi recovery was not affected by NBD-Cl− (100 µmo/L), a H+-ATPase inhibitor or Zn2+ (100 µmo/L), a voltage-activated H+ channel inhibitor (data not shown). When the JH was compared, the effect of IL1β was significantly greater than those of all the other hormones ( Fig. 1C ).
(
Effects on pHi in COC
When the experiments were repeated with the chondrocytes from osteoarthritic cartilage (COC), the effects were significantly different; the basal pHi was lower, 6.37 ± 0.24 (n = 10), and the effects of the hormones had the same trend described for CHC ( Fig. 1A ). The pHi recovery after an ammonium prepulse in these cells was attenuated (2.936 ± 0.059 mmol/L/min in CHC, n = 14 and 1.618 ± 0.173 mmol/L/min in COC, n = 8, P < 0.05) and the tested agents failed to affect it; this effect was also amiloride-sensitive and dependent on extracellular Na+ ( Fig. 1D ) and was not affected by NBD-Cl− (data not shown).
Effects on pHi Response to HTS in CHC
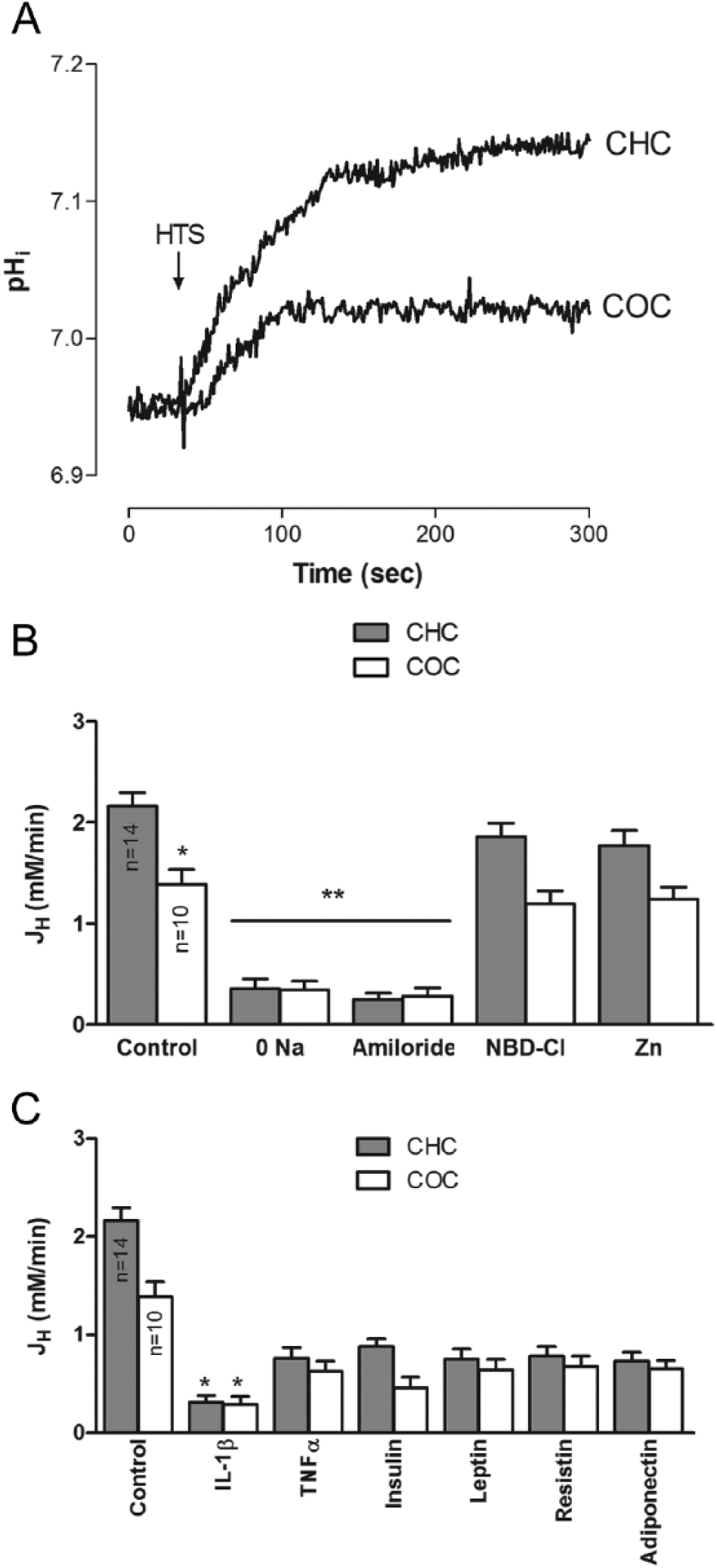
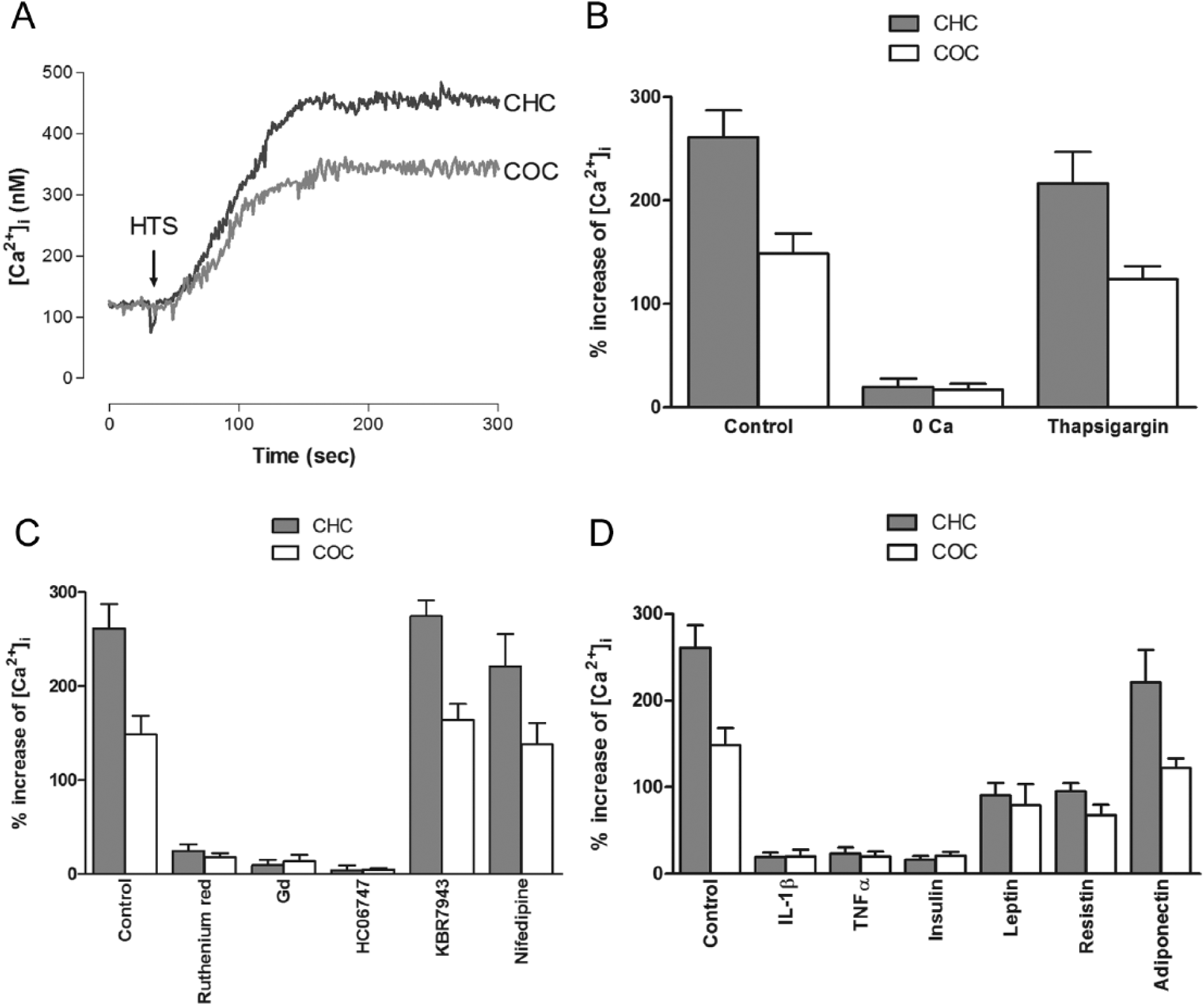
As demonstrated before in bovine chondrocytes, 35 an HTS caused a pHi increase in both CHC and COC, but the effect on the later was significantly smaller ( Fig. 2A and B ). This increase was sensitive to amiloride and dependent on extracellular Na+, but the pHi increase did not respond to treatment with NBD-Cl or Zn2+ (Figure 2B). Furthermore, all the hormones tested caused an attenuation of this response; however, the effect of IL1β was significantly greater than that of all the other factors ( Fig. 2C ).
(
Effects on [Ca2+]i in CHC
The effects of the same hormones at the same concentrations on [Ca2+]i were also evaluated over periods of 300 seconds in Fura-2-loaded CHC. In all cases, Fura-2 loading was performed prior the incubation with these agents. The control [Ca2+]i was 96.5 ± 17.2 (n = 20). Leptin, resistin, and adiponectin failed to affect basal [Ca2+]i. However, IL1β, TNFα, and insulin significantly increased [Ca2+]i after a 1-hour preincubation ( Fig. 3A ). To establish the origin of this rise in [Ca2+]i, the chondrocytes were treated with thapsigargin (1 µmol/L, 30-minute preincubation in Ca2+-free HBS) prior to the hormone treatment to deplete intracellular stores or were resuspended in Ca2+-free extracellular solution. There was a significant attenuation of the increase following treatment with each hormone in Ca2+-free extracellular solution, but thapsigargin treatment had no effect ( Fig. 3B ). Furthermore, this rise was not affected by nifedipin (1 mmol/L), a L-type voltage-activated Ca2+ channels inhibitor (LVACC); ruthenium red (10 µmol/L), a nonspecific TRPV channels inhibitor; Gd3+ (10 µmol/L), a stretch-activated channels (SAC) inhibitor, or HC-067047 (100 nmol/L), a specific TRPV4 channel inhibitor, but it was significantly attenuated by KBR7943 (50 µmol/L), a specific Na+–Ca2+ exchanger (NCX) inhibitor, in all cases ( Fig. 3D ).
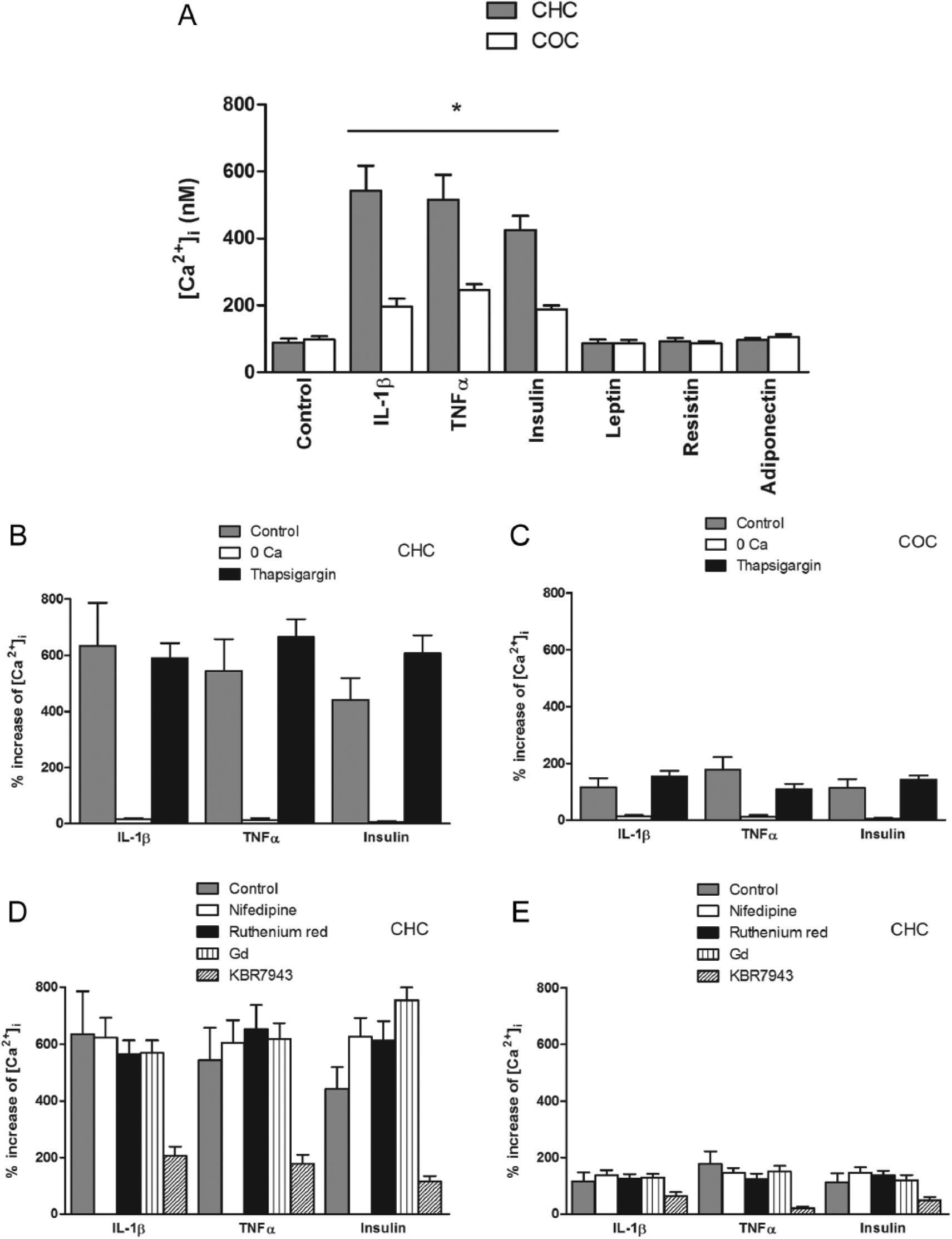
(
Effects on [Ca2+]i in COC
In COC, the basal [Ca2+]i was not significantly different from CHC, but the effects of the hormones on this parameter were different. The IL1β-, TNFα-, and insulin-induced [Ca2+]i increases were greater than those in CHC ( Fig. 3A ); these effects were dependent on extracellular Ca2+, were not affected by thapsigargin treatment ( Fig. 3C ) and were inhibited by KBR7943 but not by the other inhibitors ( Fig. 3E ). As in CHC, leptin, resistin, and adiponectin had no effect on the basal [Ca2+]i of these cells.
Effects on [Ca2+]i Response to HTS in CHC
Similar to bovine chondrocytes, 34 an HTS caused a [Ca2+]i increase in CHC ( Fig. 4A ); this effect was inhibited by ruthenium red and HC-067047, but was not affected by Gd3+, nifedipin, or KBR7943 ( Fig. 4B ), which indicates a role for TRPV4 channels in this response but not for SAC, VACC, or NCX. Furthermore, all the hormones tested, with the exception of adiponectin, attenuated the HTS-induced rise in [Ca2+]i, although the effects of leptin and resistin were to a lesser extent ( Fig. 4C ). In COC, the response was smaller compared with CHC ( Fig. 4A ) and all the hormones tested with the exception of adiponectin, inhibited the response, which was also inhibited by ruthenium red, Gd3+ and HC-067047, but were not affected by nifedipin or KBR7943 as in COC ( Fig. 4B and C ).
(
Discussion
The present study explored the effects of a number of adipokines and insulin on pHi and [Ca2+]i in human articular chondrocytes from both healthy and osteoarthritic cartilage. The present study demonstrated that IL1β affected the basal pHi, in contrast to TNFα, insulin, leptin, resistin, and adiponectin. However, all these agents did affect the pHi recovery after an ammonium prepulse, which indicates that these hormones play a role in the regulation of this parameter. Because the effect in all cases was attenuated by amiloride and was dependent on extracellular Na+, these effects can be attributed to an action on NHE, which has been shown to be one of the most significant regulators of pHi in chondrocytes and other cells. 52 Furthermore, Tattersall et al. 17 demonstrated that IL1β modulates NHE, thereby affecting the pHi recovery in bovine chondrocytes, and that this effect occurs after an acute exposure, findings that are in agreement with this study. However, there are no studies regarding the effects of other adipokines or insulin on pHi in chondrocytes or other cells. Whether the effects of these agents on NHE are direct or through other intracellular mechanisms must be addressed by further research. All these hormones have receptors in chondrocytes19,28,53,54 and have been related to the inflammatory process in which OA cartilage is immersed. 15 Chondrocyte pHi is a key factor in the regulation of cartilage matrix synthesis as demonstrated by a number of studies. 31 It is worthwhile noting that the impairment of matrix metabolism is one of the most important determinants of the development and severity of OA. 3
Hypotonic shock produced an increase in the pHi of human articular chondrocytes, which is in agreement with the findings in bovine articular chondrocytes. 35 This effect is mediated by the activation of NHE, as suggested by the inhibition by amiloride and the dependence on extracellular Na+. In contrast to bovine chondrocytes, in which voltage-activated H+ channels seems to be responsible, 35 in human articular chondrocytes, Zn2+, a H+-channel inhibitor, failed to affect the pHi increase.
Furthermore, when the effects of the tested agents on the HTS-induced pHi increase were explored, all of the agents attenuated the HTS-induced effect, with the action of IL1β being significantly greater than the others.
The pHi of chondrocytes from osteoarthritic cartilage was significantly lower than that of chondrocytes from healthy cartilage. This intracellular acidification may help to explain the impairment of matrix metabolism that is observed in osteoarthritic cartilage 55 and could reflect the alteration of the mechanisms that regulate the pHi as a consequence of the pathophysiological process of OA. Furthermore, the response of chondrocytes isolated from osteoarthritic cartilage to insulin and cytokines was significantly different. These differential responses in CHC and COC could reflect alterations in chondrocyte homeostasis as a result of OA pathophysiological process; in other words, chondrocytes living in osteoarthritic cartilage behave in a different way when compared with chondrocytes living in healthy cartilage. 3 Articular chondrocytes alkalinize in response to HTS 35 and that response may be a part of the adaptation to the osmotic challenges that these cells have to face physiologically; according to this study this response is impaired in COC. Given that pHi regulates matrix metabolism, 56 alterations in chondrocyte pHi adaptive response can alter matrix composition, leading to a dysfunctional cartilage and thus OA. According with the present study, the effects on pHi are mediated by NHE; this transporter may be regulated by a number of agents and its function can affect significantly pHi, particularly in articular chondrocytes.39,57 The way in which NHE is affected in OA needs to be determined at a molecular or functional level by further studies.
IL1β, TNFα, and insulin increased the basal [Ca2+]i, but leptin, resistin, and adiponectin had no effect on this parameter; this effect, dependent only on extracellular calcium, is mediated by the activation of NCX as suggested by the pharmacological profile of the [Ca2+]i effect, which is in agreement with our findings in bovine articular chondrocytes. 58 However, all of these agents, except for adiponectin, inhibited the HTS-induced [Ca2+]i increase, which is mediated by TRPV4 channels as indicated by the effects of specific inhibitors of this osmosensitive cation channel; the effects of these inhibitors on the HTS-induced [Ca2+]i increase occurred similarly to what was observed in bovine articular chondrocytes. 34 The fact that adiponectin had no affects whatsoever on the basal [Ca2+]i or the HTS-induced [Ca2+]i increase cannot be easily explained because this adipokine also has pro-inflammatory effects on osteoarthritic joints. Again, whether these actions were a consequence of a direct effect of these hormones on the transporter or whether they occurred secondary to the activation of other mediators requires investigation.
The [Ca2+]i response of the chondrocytes from osteoarthritic cartilage was different from the chondrocytes from healthy cartilage. Although the basal [Ca2+]i was similar in both types of cells and the implied transporters are similar, IL1β, TNFα, and insulin induced a smaller [Ca2+]i increase and a HTS produced a larger [Ca2+]i increase in CHC than in COC. Furthermore, all the hormones tested, with the exception of adiponectin, attenuated this hypotonicity-induced [Ca2+]i increase in both types of cells, but the effect was more pronounced in COC. This is another example of a dissimilar response in COC in comparison with CHC, which may help understand the role of Ca2+ homeostasis alterations in OA pathophysiology. NCX, the transporter involved in the [Ca2+]i increase induced by adipokines, and TRPV4, the pathway that was responsible for [Ca2+]i increase in response to a hypotonic challenge, may be regulated by a number of factors,59-62 which may be altered in the development of OA and could lead to an impaired function of this channel in COC; since these 2 transporters can affect [Ca2+]i, which in turn regulates matrix synthesis, a deleterious effect on cartilage function will result. However, further studies need to be conducted in order to clarify the role of NCX and TRPV4 in the OA pathophysiological process.
In sum, all of these findings indicate that the responses of cells isolated from the cartilage of a patient with OA are different from those of cells from a healthy tissue, which suggests that an alteration of pH and [Ca2+] homeostasis is part of the pathophysiological process of OA. These factors can affect matrix synthesis because intracellular ionic composition is a matrix modulator. Furthermore, the effects of adipokines and insulin revealed in the present study demonstrate that these hormones affect pH and [Ca2+] homeostasis, which may explain the association between obesity and OA at a cellular level; however, the mechanisms by which these agents modulate pHi and [Ca2+]i requires further research.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Colciencias, Bogotá, Colombia.