
Abstract
Aberrant activation of the RAS signaling pathway contributes to nearly all human cancers, including gliomas. To determine the dependence of high-grade gliomas on this signaling pathway, we developed a doxycycline-regulated KRas glioma mouse model. Using this model we previously demonstrated that inhibition of KRas expression in gliomas induced by activated KRas and Akt results in complete tumor regression. We have also shown that, in the context of Ink4a/Arf loss, abrogation of KRas signaling is sufficient to decrease tumor burden but resistance ensues. In this study, we sought to determine the effect of activated Akt signaling in combination with activated KRas and loss of Ink4a/Arf on the growth and recurrence of brain tumors following suppression of KRas expression. We observed significant tumor formation in Ink4a/Arflox/lox mice injected with retroviruses containing tetracycline responsive element (TRE)-KRas, Tet-off, Akt, and Cre. Abrogation of KRas signaling resulted in significant tumor regression; however, resistance developed after a relatively short latency. Tumor recurrence occurred more rapidly and the tumors were more aggressive in the presence of activated Akt signaling compared with loss of Ink4a/Arf alone suggesting that this pathway contributes to tumor progression in this context.
Introduction
Glioblastoma multiforme (GBM; World Health Organization Grade IV Astrocytoma) is the most common and aggressive type of brain tumor arising de novo (primary GBM) or progressing from lower grade astrocytic malignancies (secondary GBM). 1 The current standard of care for GBM is surgical resection followed by radiotherapy and temozolomide treatment. 2 However, GBM is incurable with current frontline therapies, and survival rates are among the worst of all human cancers. 3 Understanding the pathways that regulate gliomagenesis will aid in establishing more successful therapeutic agents for targeting this disease.
Recently, a comprehensive genomic characterization of human glioblastoma was performed as part of a wider coordinated effort to understand the molecular basis of cancers. 4 Multiple genetic abnormalities were found to be associated with the pathogenesis of gliomas. It was reported that 74% of tumor samples had aberrant alterations in 3 major signaling pathways: receptor tyrosine kinase (RTK) signaling and the p53/RB tumor suppressor pathways. 4 Specifically, alterations in RTK growth factor receptors (EGFR, PDGFR, MET, and ERBB2), resulting in downstream activation of RAS and AKT, were found in 88% of GBM patients. 4 RAS is a small GTPase involved in communicating mitogenic signals from outside the cell to the nucleus, which ultimately results in cell growth and division. 5 Upstream and downstream oncogenic signals and/or inherent mutations linked to RAS activation are observed in nearly all human cancers. 6
The AKT protein kinases are also critical regulators of multiple cellular functions including cell growth, survival, and metabolism. 7 AKT is a serine-threonine kinase activated by growth factor mediated signals transmitted via phosphatidylinositol 3′-kinase (PI3K). In normal cells, growth factor dependent activation of PI3K is tightly controlled by the potent tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN). Aberrant activation of AKT can result from mutations in upstream regulators (such as RTKs), overexpression of AKT, or deletions of negative regulators (such as PTEN). The PI3K/AKT pathway is often dysregulated in a wide range of cancers including GBM. 7
A significant percentage of high-grade gliomas also have homozygous deletions that functionally inactivate the cyclin-dependent kinase inhibitor 2A (CDKN2A) locus, which encodes 2 independent tumor suppressor protein products: p16INK4A and p14Arf (p19Arf in mice). 4 p16INK4a is a specific inhibitor of Cyclin D complexes, specifically CDK4 and CDK6. By inhibiting the kinase activity of these complexes, p16INK4a blocks RB phosphorylation and prevents progression from G1 to S in the cell cycle. 8 The physiological role of p14ARF appears to involve stabilization of p53 levels, through the inhibition of MDM2, and cell cycle arrest in response to oncogenic stimuli. 8 Disruption of the RB and p53 pathways are found in 87% and 78% of GBM, respectively, demonstrating a fundamental requirement for the deletion of these tumor suppressor pathways in gliomagenesis. 4
GBM tumors comprise a heterogeneous population of aberrant cells making them extremely difficult to treat. Determining which genetic mutations are drivers of gliomagenesis, as opposed to “passenger” mutations, is essential in delineating successful therapeutic treatments. One approach for determining the role of such genetic mutations is to use mouse models. We and others have successfully used a series of replication-competent retroviral vectors based on Rous Sarcoma Virus (RSV), a member of the Avian Leukosis Virus (ALV) family, to study the roles of different genes in brain tumor initiation and progression (reviewed in Li et al 9 and von Werder et al 10 ). This system uses a replication competent, ALV LTR, containing a splice acceptor, Bryan Polymerase viral vector, termed RCASBP(A). 9 The major advantage of these retroviral vectors is their ability to deliver genes efficiently and stably to somatic cells that express the TVA receptor. In this model, expression of the TVA receptor is driven from the nestin promoter, which is expressed in precursors of neural and glial cells. Using this approach, we have shown that suppression of KRas expression in glioblastomas induced by activated KRas and Akt results in rapid tumor regression and significantly increased survival. 11 Moreover, resistance did not develop in any of these mice throughout the duration of the study. This demonstrates that sustained activation of the Ras pathway is required for tumor maintenance in the context of activated Akt. We have further shown that in the context of Ink4a/Arf loss, inhibition of KRas is also sufficient to significantly decrease tumor burden. 12 However, tumors became resistant in half of the mice in this context, suggesting that Ink4a/Arf loss enhances the emergence of recurrent tumors. 12 In this study, we sought to extend our current findings and examine the role of Akt signaling, in combination with activated KRas and loss of Ink4a/Arf, on the growth and recurrence of brain tumors following suppression of KRas expression. Despite the initial dependency of these primary gliomas on continued KRas signaling, tumors progressed to a more aggressive, KRas-independent, high-grade glioma in 70% of the mice. Recurrence occurred more rapidly and tumors were more aggressive in the presence of activated Akt as compared with loss of Ink4a/Arf alone, suggesting that this pathway contributes to tumor progression in this context. Our findings have important implications for the design of therapeutic strategies aimed at eliminating this disease.
Results
High penetrance of glioma formation initiated by expression of Akt and conditional KRas in the context of Ink4a/Arf deficiency
In this study, we extended our observations to examine the role of activated Akt signaling following KRas inhibition in the context of Ink4a/Arf deficiency. In Ink4a/Arflox/lox mice, the lox sites flank exons 2 and 3 of the Ink4a/Arf locus such that Cre-mediated excision eliminates both p16 ink4a and p19 Arf . In the absence of Cre, homozygous Ink4a/Arflox/lox mice and cells isolated from these mice express normal levels of p16 ink4a and p19Arf. 13 Intracranial injection of TRE-KRas, Tet-off, Akt, and Cre containing retroviruses into newborn Nestin-TVA;Ink4a/Arflox/lox mice resulted in tumor formation in greater than 80% of injected mice (Fig. 1A). Importantly, TVA-negative;Ink4a/Arflox/lox control mice infected with the same viruses remained tumor free (P = 4.8 × 10−7; Fig. 1A). It has previously been established that infection with KRas, Akt, or Cre containing viruses alone, or the combination of Akt and Cre containing viruses, is insufficient for tumorigenesis in these mice.14,15 There is also no significant incidence of tumor formation in Ink4a/Arf-null mice within 100 days of age.16,17 Thus, delivery of these viruses in combination results in highly penetrant glioma development in this model.
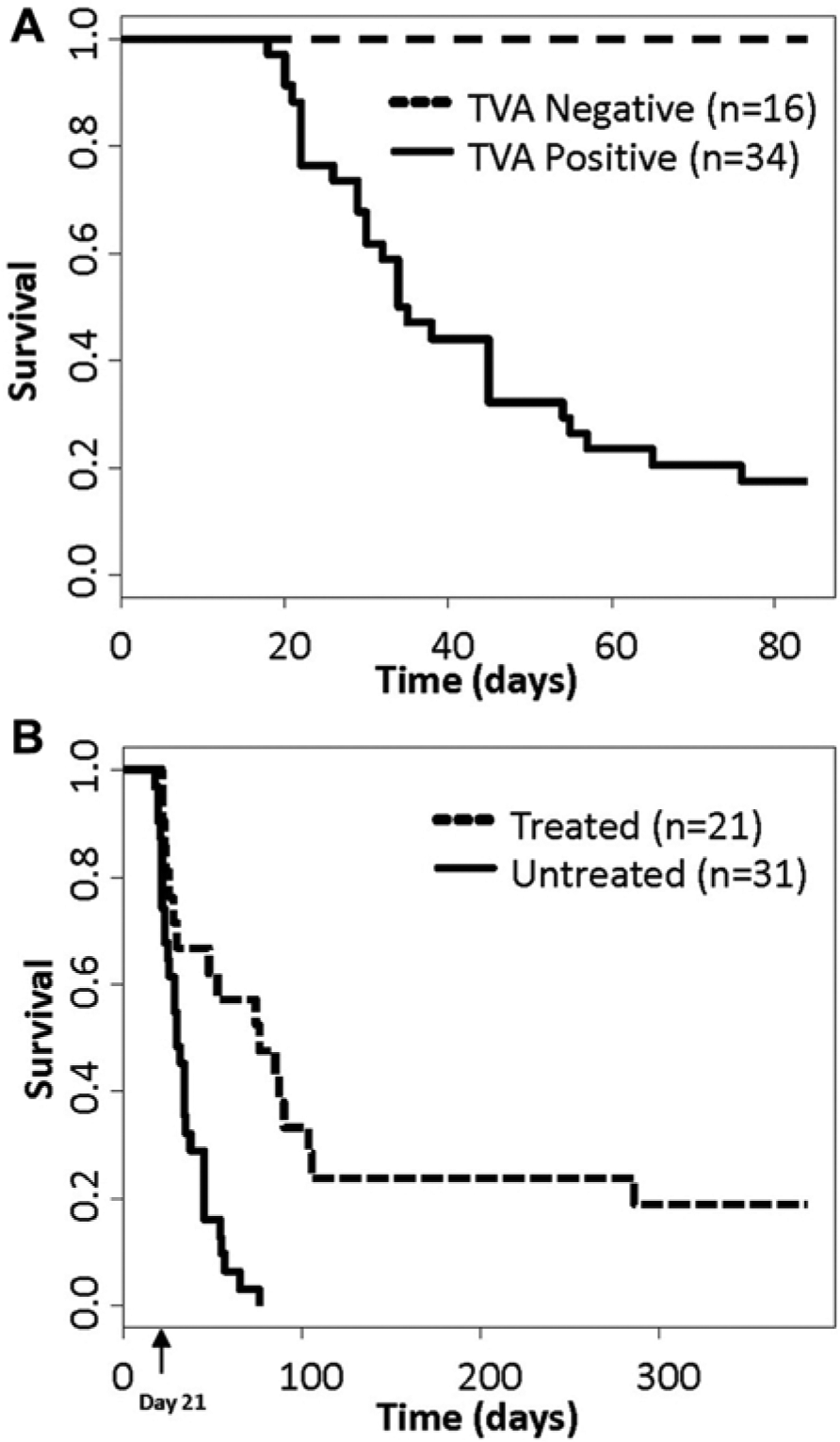
Kaplan-Meier percent survival curves. All mice were injected with TRE-KRas, Tet-off, Akt, and Cre containing viruses at birth. (
Suppression of KRas expression increases survival
To determine whether the suppression of KRas signaling in KRas and Akt induced gliomas in the absence of Ink4a/Arf expression results in tumor regression, tumors were induced in Nestin-TVA;Ink4a/Arflox/lox mice by intracranial injection of TRE-KRas, Tet-off, Akt, and Cre containing retroviruses at birth. Magnetic resonance imaging (MRI) was used to identify tumor-bearing mice and to monitor tumor growth longitudinally throughout the study. Dox was administered through the food to one cohort of tumor bearing mice at 21 days of age to suppress KRas expression while a separate cohort of tumor bearing mice received standard feed (Fig. 1B). Survival rates were compared between untreated mice and mice given Dox to determine whether the loss of KRas expression increased survival in this context. While the median survival for the untreated mice was 30 days, the median survival for the treated mice was 76 days (P = 5.1 × 10−5) (Fig. 1B). On treatment with Dox, tumor regression was observed by MRI in 14 out of the 21 animals; the remaining 7 died within the first week of treatment. MRI data from a representative mouse that showed a complete response while on Dox treatment is shown in Figure 2. A tumor was visible pre-Dox treatment (Fig. 2A and B) while no tumor was detected at 19 weeks of Dox treatment (Fig. 2C) or 1 year of Dox treatment (Fig. 2D), which was the experimental endpoint. We have previously demonstrated that Dox has no innate tumor suppressive properties 18 ; therefore, tumor regression is due to inhibition of KRas expression.
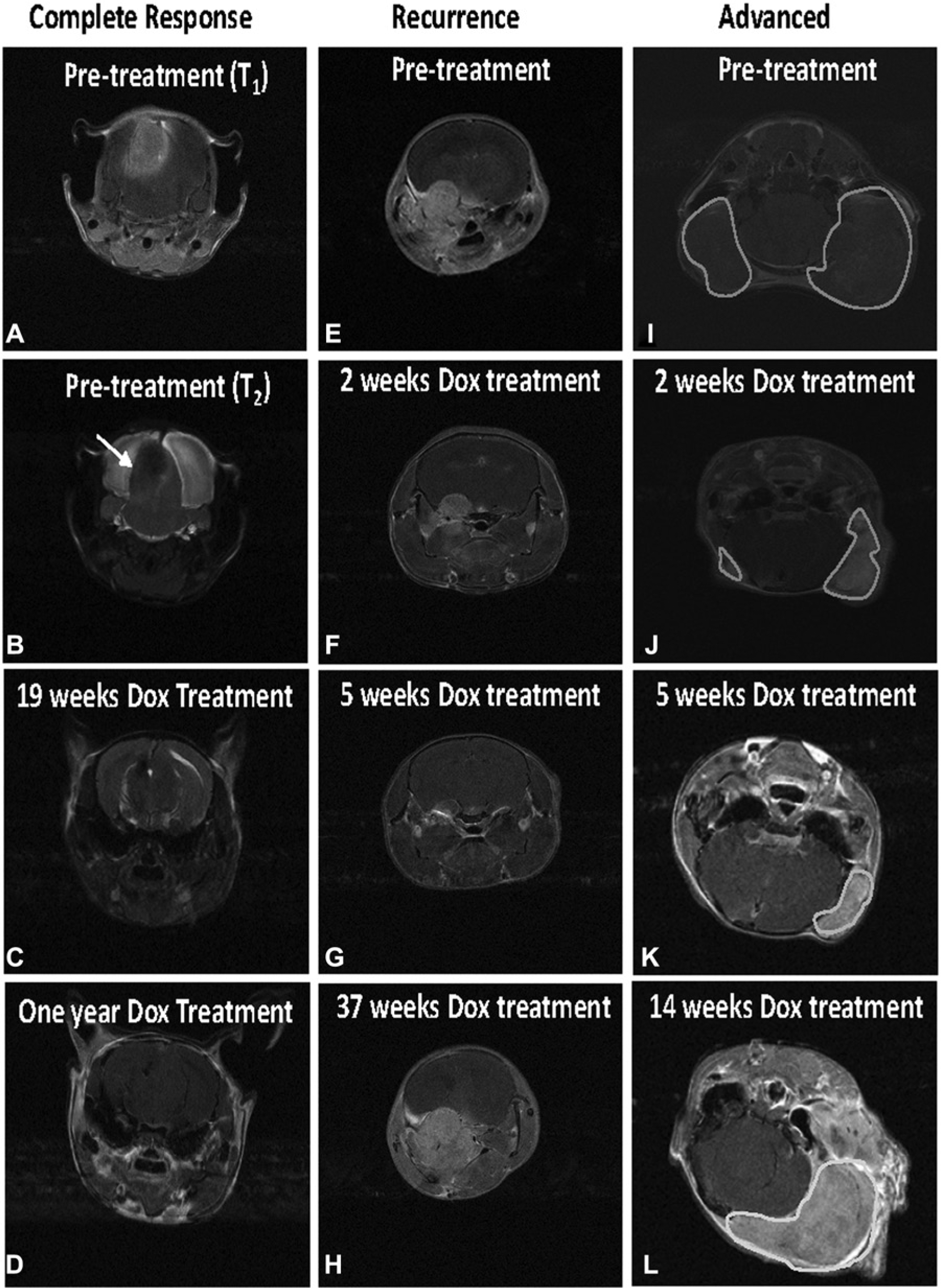
Magnetic resonance imaging (MRI) analysis of tumor-bearing mice injected with TRE-KRas, Tet-off, Akt, and Cre viruses. Mice were anesthetized with isoflurane and images acquired on a 7-T Bruker MRI using T1-weighted multi-spin–multi-echo (MSME) or axial T2-weighted TurboRARE sequences in the presence of gadolinium-based contrast agent before treatment and at the indicated times of Dox treatment. All images are T1-weighted unless otherwise indicated. (
Recurrent tumors develop in the presence of KRas inhibition
Of the 14 mice that demonstrated tumor regression following Dox treatment, 10 showed rapid relapse while the remaining 4 demonstrated complete remission while on Dox for 1 year. Interestingly, in 9 of the 10 mice that developed recurrence, tumors grew back nearly simultaneously after a mean latency of 78 ± 6 days. The tenth mouse experienced tumor recurrence after a latency of 261 days (Fig. 1B). The MRI data from this mouse revealed a tumor pre-Dox treatment (Fig. 2E), which was significantly reduced following just 2 weeks of Dox treatment (Fig. 2F). The tumor was barely detectable at 5 weeks of Dox treatment (Fig. 2G); however, a large tumor was visible at 37 weeks of Dox treatment (Fig. 2H). These tumors recurred while the mice were on Dox treatment in the absence of virally delivered KRas expression.
Since many human gliomas are diagnosed at an advanced size, one cohort of tumor-bearing mice was treated with Dox once the tumors were more established. MRI data from a representative mouse is shown in Figure 2. Two tumors were visible pre-Dox treatment (Fig. 2I) and within 2 weeks of Dox-treatment significant tumor regression was observed in both tumors (Fig. 2J). The tumors continued to regress for an additional 5 weeks at which point the smaller tumor was no longer detectable (Fig. 2K). The other tumor remained detectable by MRI but was significantly reduced compared to its tumor size pretreatment (Fig. 2K). However, significant growth of this tumor was observed at 14 weeks even in the presence of continuous Dox treatment (Fig. 2L).
Immunohistochemical analysis confirms expression of virally delivered genes and tight regulation of KRas expression
Brain tissue from all mice was analyzed histologically to detect expression of the virally delivered genes and confirm Dox-mediated regulation of KRas expression. We have previously demonstrated that KRas expression is significantly reduced within 3 days of Dox treatment. 12 Representative sections from a primary untreated tumor and a recurrent tumor that had been treated with Dox for 67 days are shown in Figure 3. Sections were stained with H&E to provide structural comparison and images containing both tumor and normal tissue are shown (Fig. 3A and B). Akt expression was visualized by immunohistochemical (IHC) staining of the tumor sections with antibodies to the HA-epitope tag on virally delivered Akt in both untreated and Dox-treated tumors as expected (Fig. 3C and D). IHC analysis of brain tissue confirmed KRas expression in untreated cohorts and demonstrated that KRas expression was reduced, as expected, in the Dox-treated mice (Fig. 3E and F). Cre activity was also confirmed by loss of p19Arf expression in the tumor tissue (Fig. 3G and H). Adjacent normal tissue served as a positive control for p19Arf staining.
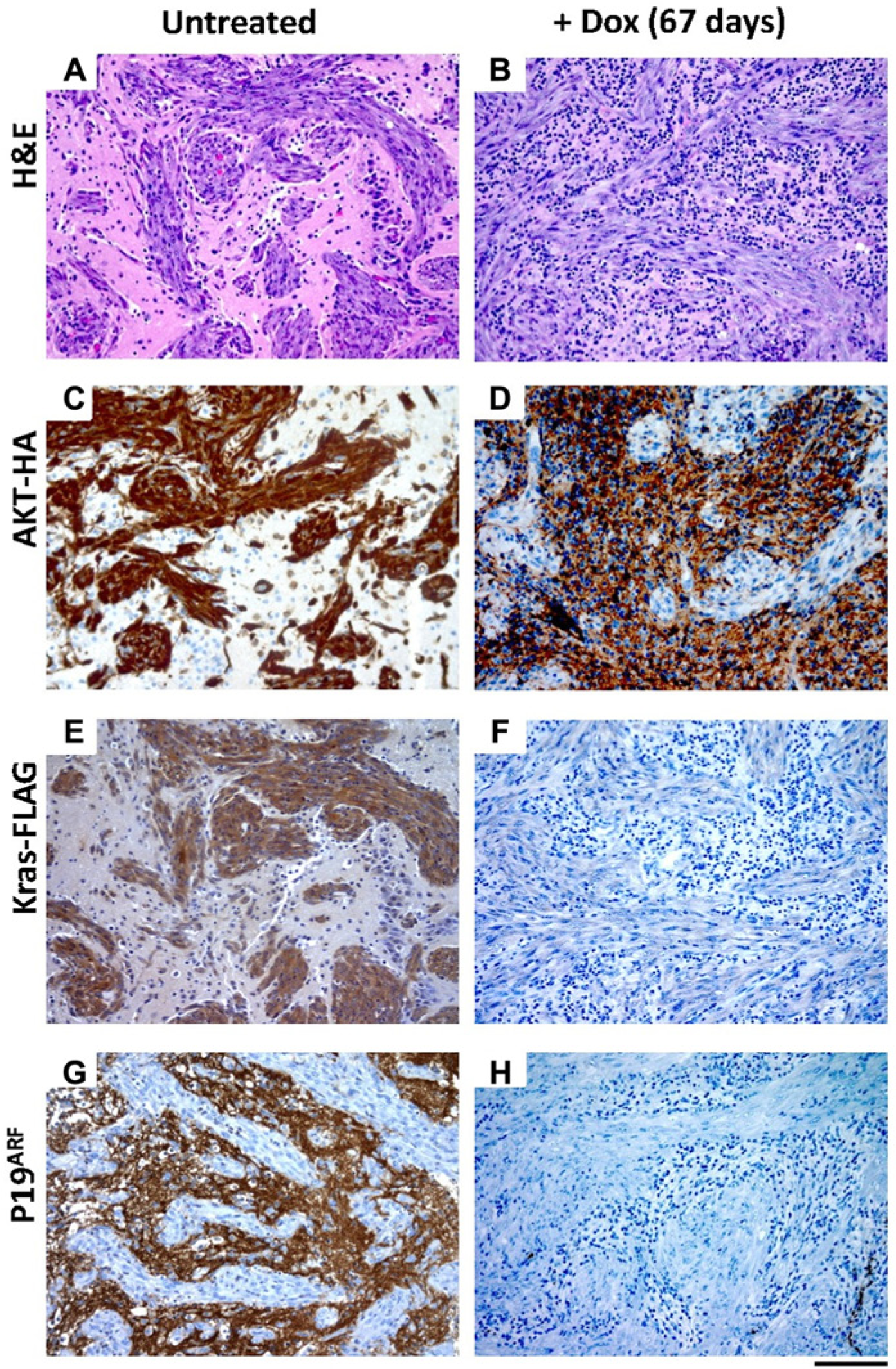
Immunohistochemical characterization of a recurrent glioma arising in N-TVA;Ink4a/Arflox/lox mice injected with TRE-KRas, Tet-off, Akt, and Cre containing viruses. All images are serial sections from the same mouse sacrificed at 21 days of age with no treatment (
The presence of activated Akt leads to more aggressive tumors and shorter latency to recurrence
Both primary and recurrent tumors induced with TRE-KRas, Tet-off, Akt, and Cre were evaluated histologically and compared to primary and recurrent tumors induced with TRE-KRas, Tet-off, and Cre in the absence of activated Akt (Fig. 4A-H). The majority of primary tumors initiated by activated KRas and Akt in the context of Ink4a/Arf loss resembled highly invasive GBM (Fig. 4A-D). Primary tumors were polymorphic, possessed a diffuse growth pattern, and contained several areas of pseudopalisading necrosis (Fig. 4A and B). While the recurrent tumors were less necrotic than the primary tumors, they remained extremely polymorphic with highly condensed heterochromatin (Fig. 4C and D). The recurrent tumors also showed pronounced angiogenesis in the context of activated Akt compared to recurrent tumors established from expression of TRE-KRas, Tet-off, and Cre (compare Fig. 4C-D with G-H). The polymorphism is likely a result of Akt expression since tumors initiated with TRE-KRas, Tet-off, and Cre in the absence of activated Akt were histologically distinct (Fig. 4E-H). Tumors that recurred in the absence of Akt appeared less aggressive and were more homogenous (Fig. 4G and H), suggesting that the presence of activated Akt contributes to a more aggressive GBM phenotype (Fig. 4C, D, G, and H). Comparison of survival rates among the Akt or non-Akt containing cohorts of mice that developed recurrent tumors while on Dox treatment revealed a significant difference; the presence of Akt was associated with more rapid recurrence and decreased survival compared to those mice with tumors that lacked Akt (Fig. 5).
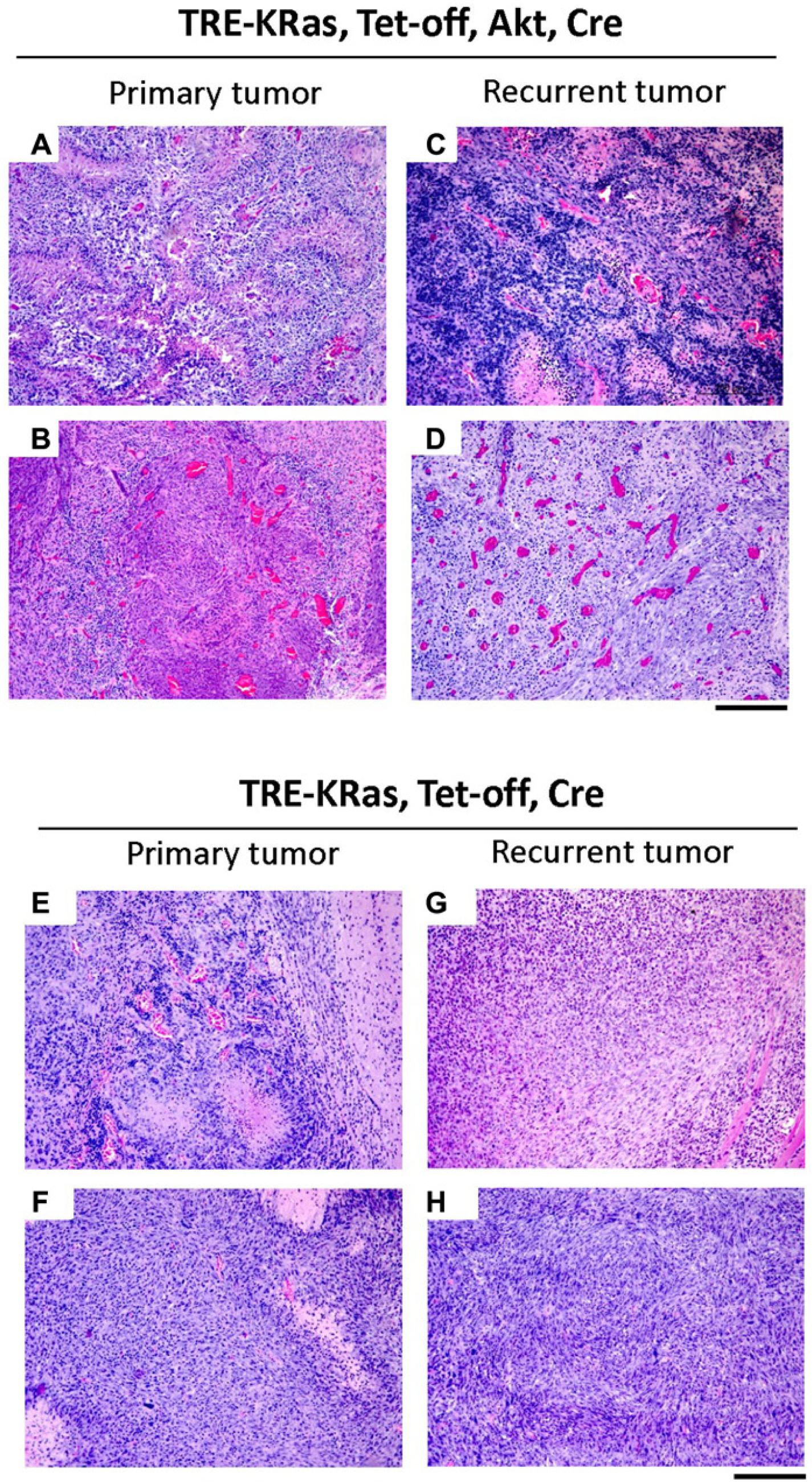
Comparison of primary and recurrent tumors in the presence or absence of activated Akt. Histological comparison of gliomas induced in N-TVA;Ink4a/Arflox/lox mice after injection with TRE-KRas, Tet-off, and Cre containing viruses (
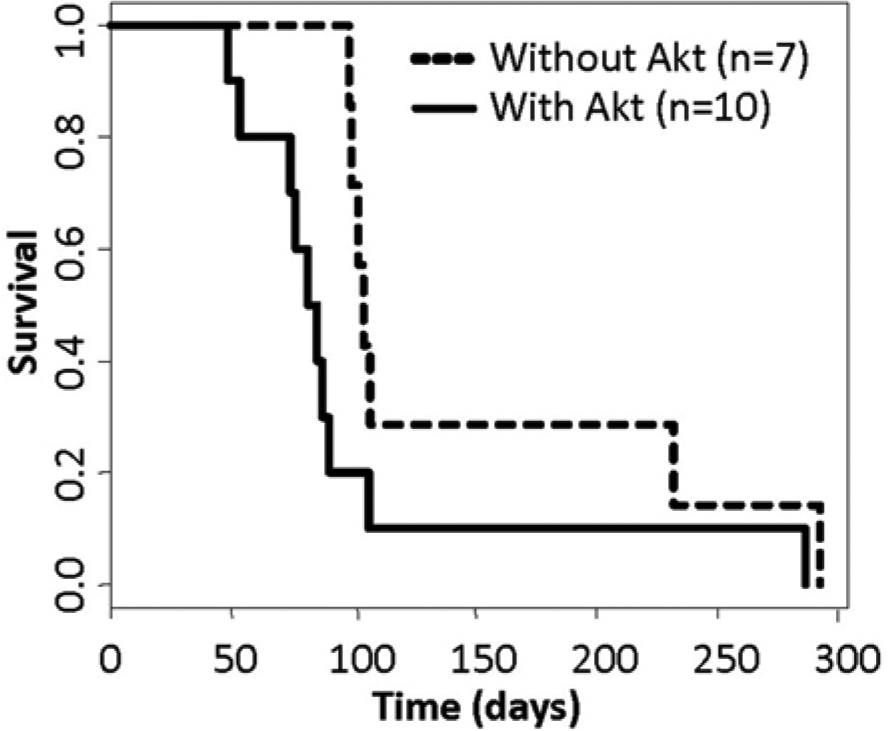
Kaplan–Meier percent survival curves. Comparison of survival rates among the mice that developed recurrent tumors while on Dox treatment either in the presence (n = 10; solid line) or absence of Akt (n = 7; dashed line) revealed a significant difference, P = 0.05. Doxycycline treatment was continuous for the duration of the experiment.
Discussion
It has previously been shown that KRas expression combined with either Akt activation or deletion of Ink4a/Arf leads to the development of high-grade gliomas in mice.14,19 We extended these findings to determine the role of KRas signaling in tumor maintenance by using a Tet-regulated KRas viral vector. We observed complete regression in brain tumors induced by activated KRas and Akt following abrogation of KRas signaling. 11 In addition, gliomas induced by activated KRas in the context of Ink4a/Arf loss showed significant tumor regression on treatment with Dox; however, a significant number of these tumors recurred suggesting that Ink4a/Arf loss promotes tumor recurrence following KRas inhibition. 12 This Tet-regulated glioma model has, therefore, been essential in providing insights into the role of KRas signaling in glioma maintenance. In this study, we evaluated the effect of Akt activation on tumor recurrence in the context of activated KRas and Ink4a/Arf loss following suppression of KRas expression. We observed that KRas inhibition was sufficient for tumor regression but resistance ensued in the majority of mice. Tumor recurrence occurred more rapidly and tumors were more aggressive in the context of activated Akt and Ink4a/Arf loss, compared with Ink4a/Arf loss alone, suggesting that activation of Akt signaling contributes to tumor growth in this context. Together these findings demonstrate that the effectiveness of inhibiting a single target, such as KRas, as a therapeutic approach is highly dependent on the combination of genetic events present within the tumor.
PI3K/Akt pathway activation is known to mediate resistance to MAPK inhibition in RAS mutant cancers,20,21 and accordingly we have observed greater resistance to MEK inhibition in tumor cells with higher levels of PI3K/AKT signaling. 22 We have previously demonstrated that combined inhibition of MEK and PI3K/mTOR induces a significant amount of cell death in a large panel of human glioma cell lines. Importantly, enhanced cell death was observed with combined inhibition of MEK and PI3K/mTOR in nearly all of the glioma cell lines regardless of the level of Akt activity present. 22
The Akt protein kinases are critical regulators of multiple cellular functions while loss of Ink4a/Arf largely leads to deregulated cellular growth signaling pathways. Together, these alterations confer a survival advantage to cells by turning off key tumor suppressor pathways while enabling oncogenic transformation. Thus, the combination of either Ink4a/Arf loss and activated KRas or Ink4a/Arf loss, activated Akt and KRas, appear to reduce the susceptibility of gliomas to inhibition of KRas signaling. 12 However, the combination of activated Akt and KRas in the presence of Ink4a/Arf expression was susceptible to targeted KRas inhibition and recurrence was not observed in this context. 11 Our findings suggest that tumor recurrence is a common event in Ras/MAPK-driven tumors in the context of Ink4a/Arf loss and activated Akt, both of which commonly occur in human gliomas. Highly effective inhibitors targeting several key components of the Ras/MAPK and PI3K/Akt pathways have been developed and many of these are in clinical trials for gliomas (reviewed in Chappell et al 20 ). This mouse model is thus key to deciphering the genetic combinations that affect the efficacy of targeted therapies as well as the potential for recurrence.
It is unclear how gliomas, formed following viral delivery of TRE-KRas, Tet-off, Akt, and Cre, are able to evade the abrogation of KRas signaling to restore tumor growth. Our data demonstrate that tumor recurrence is largely simultaneous, suggesting that the regressed tumor undergoes an additional genetic alteration that provides it with a proliferative and/or survival advantage. This survival advantage directly correlates with the combination of Ink4a/Arf loss and activated Akt. Thus, future studies will examine these recurrent tumors to identify the genetic events associated with tumor progression in this context.
Materials and Methods
Mice and genotyping
All experiments were performed in compliance with the “Care and Use of Animals” 23 and were approved by the IACUC prior to experimentation. Nestin-TVA;Ink4a/Arflox/lox mice have been described. 15 DNA was prepared from tail biopsies and genotyped by PCR for the TVA transgene and Ink4a/Arf wild-type and floxed alleles as previously described. 15
Viral constructs
The retroviral vectors used in this study are replication-competent avian leukosis virus (ALV) long terminal repeat (LTR), splice acceptor, and Bryan Polymerase-containing vectors of envelope subgroup A [designated RCASBP(A)] or RCANBP(A), which lacks the splice acceptor. The generation of RCASBP(A)Tet-off, RCANBP(A)TRE-KRas, RCASBP(A)Akt, and RCASBP(A)Cre have been described previously.11,15
Viral production
To propagate RCASBP(A)/RCANBP(A) viruses, genomic clones of the viral vectors were transfected into DF-1 cells using the calcium phosphate transfection method. 24 RCASBP(A) and RCANBP(A) viral vectors are replication-competent in the DF-1 cell line, an immortalized chicken fibroblast line, and high titer viral stocks can be obtained. 25 Viral titers were determined by assaying 10-fold serial dilutions of the viral supernatants on DF-1 cells by ELISA as previously described. 26 Virus stocks were generated from cell supernatants. The supernatants were cleared of cellular debris by centrifugation at 2,000 × g for 10 minutes at 4°C, filtered through a 0.45-µm filter, and stored in aliquots at −80°C. Virus was determined to be replication-competent by a reading of 0.200 or greater on ELISA for the viral capsid protein.
In vivo infection
Transfected DF-1 cells from a confluent culture in a 10-cm dish were trypsinized, pelleted, resuspended in 50 µL phosphate-buffered saline and placed on ice. Newborn mice were injected intracranially 2 mm ventral from bregma (intersection of the coronal and sagittal sutures) with 5 µL of infected DF-1 cells using a gas-tight Hamilton Syringe. All injected mice were monitored daily for health.
Histological analysis
Brain tissue from injected mice was fixed in formalin overnight, cut into sections, and dehydrated through a graded alcohol series in a Microm STP 420D tissue processor (Thermo Fisher, Kalamazoo, MI). Tissues were paraffin embedded and 5 µm sections were adhered to glass slides. Sections were stained with H&E or left unstained for IHC. Images were captured using a Zeiss Axio microscope equipped with an AxioCam ICc3 camera (Zeiss, Thornwood, NY).
Immunohistochemistry
Tissue sections were deparaffinized and antigen retrieval was performed in “Diva Decloaking” buffer (Biocare Medical, Concord, CA) by boiling for 10 minutes in a Decloaking Chamber (Biocare Medical). Sections were treated with 3% hydrogen peroxide and blocked in Background Sniper (Biocare Medical) for 10 minutes. Primary antibodies were diluted in Renaissance background reducing diluent (Biocare Medical). Sections were incubated overnight at 4°C and probed with Mach 4 rabbit polymer reagent (Biocare Medical) or Mach 4 mouse probe for 15 minutes followed by Mach 4 polymer for 15 minutes for mouse monoclonal antibodies. Visualization was carried out with DAB (Biocare Medical). Sections were counterstained with hematoxylin. KRas expression was detected using an antibody to the FLAG epitope (F7425, Sigma, St Louis, MO), diluted 1:200. Akt expression was detected using an antibody to the HA epitope (HA.11, Covance, Princeton, NJ), diluted 1:1000. Cre activity was assessed using an antibody recognizing p19ARF (ab80, Abcam, Cambridge, England), diluted 1:200. Cell proliferation was detected using an antibody to Ki67 (M7246, Dako, Glostrup, Denmark), diluted 1:50. Apoptosis was detected by TUNEL staining using the in situ cell death detection kit (Roche, IN) per the manufacturer’s specifications.
Magnetic resonance imaging
Mice were anesthetized via inhalation of 2% Attane isoflurane (Minrad, Orchard Park, NY) with oxygen (1.0 L/min) for 5 minutes prior to imaging and throughout the procedure. The mice were imaged using a Bruker 7 T USR 20 cm diameter bore (Model 70/20) Biospin MRI running Paravision 4. The 7 T MRI is equipped with a 6 cm inner diameter unshielded gradient coil insert (Model BGA 06) and a 35 mm inner diameter quadrature RF Birdcage coil. The mice were kept warm using a rodent (warm air) heater system (SA Instruments Inc., Stony Brook, NY). Mouse respiration was monitored using an MR-compatible monitoring and gating system (Model 1025; SA Instruments Inc.). A 50 µL bolus of MultiHance (gadobenate dimeglumine; Bracco Diagnostics Inc., Princeton, NJ) MRI contrast was delivered by intraperitoneal (IP) injection immediately prior to imaging. The imaging protocol consists of a fast gradient-echo 3-plane localizer for graphical prescription (field of view [FOV]: 4 cm; slice thickness: 1 mm; matrix: 128 × 128; TR: 100 ms; TE: 6 ms; flip angle [FA]: 30°; scan time: 13 seconds). The presence of the chelated gadolinium MR contrast was verified using an axial T1-weighted Rapid Acquisition Relaxation Enhanced (RARE) spin-echo sequence (FOV: 2.65 cm; slice thickness: 0.75 mm; matrix: 256 × 256; TR: 800 ms; TE: 7.5 ms; rare partitions: 4; scan time: 38 seconds). Quality contrast-enhanced structural images were acquired using an axial T1-weighted Multi-Slice Multi-Echo (MSME) spin-echo sequence to visualize the tumor volume (FOV: 2.65 cm; slice thickness: 0.75 mm; matrix: 256 × 256; TR: 700 ms; TE: 10.7 ms; scan time: 3 minutes). Additional structural images were acquired using an axial T2-weighted TurboRARE sequence to detect edema (FOV: 2.65 cm; slice thickness: 0.75 mm; matrix: 256 × 256; TR: 4200 ms; TE: 36 ms; rare factor: 8; scan time: 100 seconds). Fifteen to twenty 0.75 mm thick slices were typically required to cover the brain. MRIcro (http://www.mccauslandcenter.sc.edu/CRNL/) was used to convert the Bruker DICOM image files to Analyze format, and manually draw volumes of interest (VOIs) around the tumors. A Matlab (Mathworks, Natick, MA) program was written to calculate the tumor volume based on the manually drawn VOIs.
Statistical analysis
Censored survival data were analyzed using a log-rank test of the Kaplan–Meier estimate of survival.
Footnotes
Acknowledgements
We thank the Huntsman Cancer Institute mouse, imaging, and histology cores for assistance.
Authors’ Note
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the funding agency.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Huntsman Cancer Institute, the National Brain Tumor Foundation, the American Cancer Society RSG-06-198-01-TBE, and by NIH/NINDS R01 NS073870 (SLH).