
Abstract
Ras signals through both mitogenic and stress pathways and studies of Ras regulatory effects of stress pathways hold great promise to control Ras-dependent malignancies. Our previous work showed Ras activation of a stress kinase (MAPK-activated protein kinase 2 [MK2]), and here, we examine regulatory effects of MK2 on Ras oncogenesis. MK2 knockout was shown to increase Ras transformation in mouse embryonic fibroblasts (MEFs) in vitro and to enhance the resultant tumor growth in mice, indicating a tumor suppressor activity. In Ras-dependent and -independent human colon cancer, however, MK2-forced expression increases and MK2 depletion decreases the malignant growth, suggesting its oncogenic activity. The oncogenic activity of MK2 couples with its activation by both stress and mitogenic signals through extracellular signal–regulated kinase and p38α pathways, whereas its tumor-suppressing effect links to its stimulation only by stress downstream of p38α. Of interest, MK2 was shown to decrease intracellular levels of reactive oxygen species (ROS) in MEFs but increase its production in human colon cancer cells, and experiments with antioxidants revealed that ROS is required for Ras oncogenesis in both systems. These results indicate that MK2 can increase or decrease Ras oncogenesis dependent of its ROS regulatory activities.
Introduction
Ras is the most established oncogene and plays a key role in both experimental transformation and human tumor pathogenesis. 1 Experimental evidence suggests that in addition to mitogenic pathways, stress signaling plays a critical role in determining Ras oncogene activity through cross-talk and feedback mechanisms. 2 Although much has been learned from Ras-induced experimental tumors about roles of stress pathways, their application potentials in human cancer remain mostly unknown. 3
p38 mitogen-activated protein kinases (MAPKs) are major pathways that, together with extracellular signal–regulated kinases (ERKs) and c-Jun N-terminal kinases (JNKs), play a role in converting various extracellular signals into a cellular response. The p38 family consists of 4 isoforms (α, β, γ, and δ) in which p38α is ubiquitously expressed and involved in the regulation of cytokine signaling and inflammatory response, whereas functions of other p38 family proteins are just being revealed.2,4 In response to upstream signals, p38α is phosphorylated downstream of MAPK kinase 6 (MKK6) and/or MKK3, which then activates its substrates MAPK-activated protein kinase 2 (MK2) and p38-related/activated protein kinase (PRAK) and/or other proteins, leading to a biological response. 4 Our earlier study showed that p38α inhibits Ras-induced proliferation in mouse NIH 3T3 cells, 5 which has been further confirmed by knocking out MKK3/6, 6 p38α, 7 or PRAK 8 in mice, indicating that the entire p38α pathway acts as a tumor suppressor. Although MK2 is a direct substrate of p38α/β, 9 its effects on Ras tumorigenesis have not been established.
MK2 was originally cloned as a kinase downstream of ERK pathways, 10 and substantial evidence later showed that MK2 primarily acts as a p38-activated stress kinase. 9 In this report, we showed that MK2 knockout increases Ras transformation and tumorigenesis in experimental mouse cells in vitro and in vivo. Of interest, experiments with both MK2 overexpression and depletion showed that MK2 is required for the malignant growth of human colon cancer cells. The oncogenic activity of MK2 was found to couple with its stimulating effect on reactive oxygen species (ROS) production and with its activation by both stress and mitogenic signals downstream of p38/ERK pathways. The tumor suppressor activity, on the other hand, links to its stimulation only by stress signaling and its inhibition of oncogenic ROS accumulation. These results together indicate that MK2 can inhibit or promote Ras oncogenesis depending on its regulatory effects on ROS production.
Results
MK2 inhibits Ras tumorigenesis in mouse fibroblasts
To study the roles of MK2 in Ras transformation, wild-type (WT) MK2 (MK2+/+) and knockout (KO) MK2 (MK2−/−) mouse embryonic fibroblasts (MEFs) 11 were stably expressed with hemagglutinin (HA)–tagged oncogenic H-Ras through retroviral infection and analyzed for anchorage-independent growth by soft agar assays. 12 In agreement with the inhibitory role of MK2 in Ras-induced DNA synthesis in mouse NIH 3T3 cells, 5 MK2 KO significantly increases the soft agar growth, indicating its tumor suppressor activity (Fig. 1A). The increased growth is a direct effect of MK2 deletion, as it was in part rescued by the MK2 restoration (Fig. 1B).
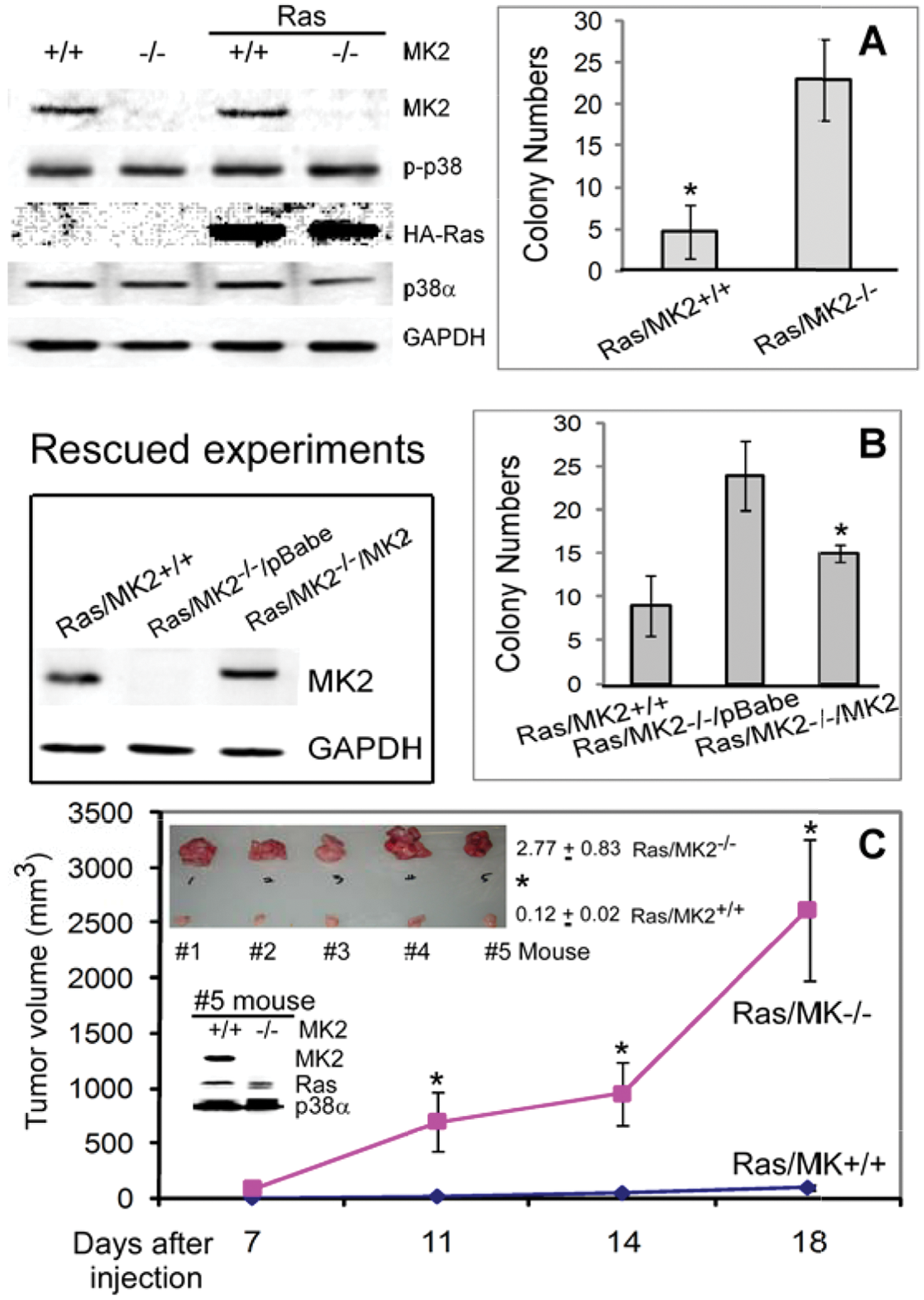
MK2 inhibits Ras oncogenesis in MEFs in vitro and in vivo. (
To further demonstrate the inhibitory role of MK2 in Ras tumorigenesis, Ras-transformed WT and KO MK2 MEFs were injected into both front flanks of nude mice, and their tumor-forming activity was then monitored. Results in Figure 1C showed that Ras-transformed MK2−/− cells gave rise to a larger tumor at all time points examined. Moreover, there is also a significant increase in tumor weights in the Ras/MK2−/− group from all 5 mice (Fig. 1C, top insert for tumor weights and bottom insert for MK2 protein expression). These results together reveal a suppressor role of MK2 in Ras tumorigenesis in MEFs in vitro and in vivo.
MK2 increases Ras-dependent and -independent malignant growth in human colon cancer cells
To examine the application potentials of MK2 as a tumor suppressor, human colon cancer HCT116 and SW480 cells were next included in our analyses. K-Ras mutation/activation is a frequent event in human colon cancer, 13 and both HCT116 and SW480 cells harbor K-Ras mutations that have been previously shown to be functionally required for their malignant growth.14,15 Although Ras-dependent colon cancer growth may not entirely mimic the Ras transformation process in MEFs, it may be one of the achievable approaches for verifying experimental tumor results in human cancer. In this regard, endogenous MK2 was first depleted by siRNA, 12 and its effects on the soft agar growth were analyzed. To our surprise, MK2 depletion inhibits the colony formation in both cell lines, indicating its oncogenic activity (Fig. 2A and 2B). Moreover, stable expression of a constitutively active MK216 also increases the growth (Fig. 2A and 2B). To demonstrate if the oncogenic effect of MK2 depends on Ras mutation, WT K-Ras expressing Caco2 human colon cancer cells was next included in the analysis. Results in Figure 2C and 2D showed a similar stimulation and inhibition of the growth by MK2 expression and knockdown, respectively. Together, these results indicate a growth-promoting role of MK2 in both Ras-dependent and -independent human colon cancer cells.
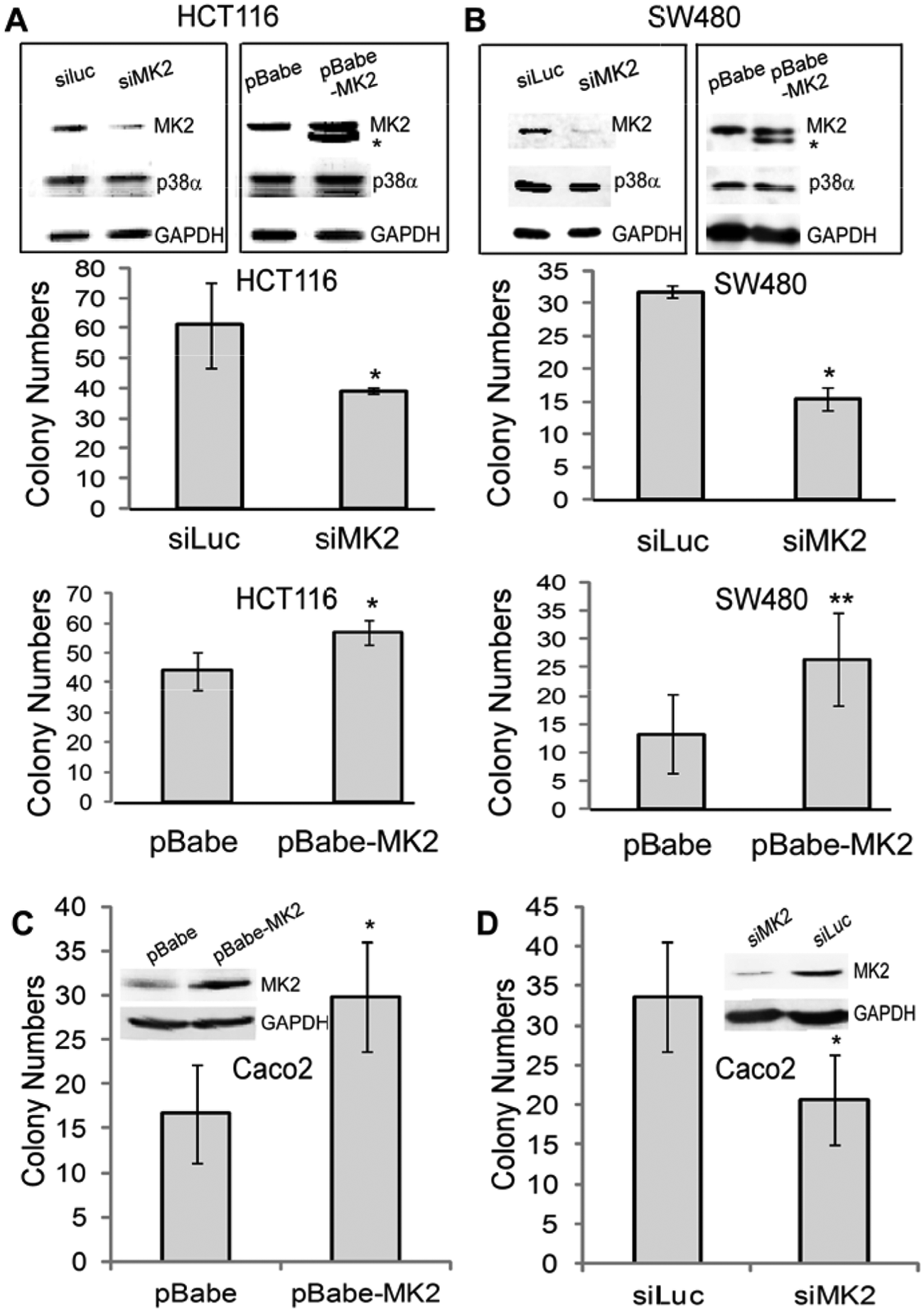
MK2 promotes the malignant growth of human colon cancer cells independent of Ras mutation/activation. (
MK2 is activated by both mitogenic and stress stimuli downstream of ERK and p38α pathways in human cells
MK2 is activated/phosphorylated at the same sites by ERK1/2 and p38α, albeit most in vivo studies only focused on its signaling downstream of p38 pathways. 9 Along with this property, cellular experiments showed that MK2 is activated only through p38 pathways in cardiac myocytes, 17 through both p38 and ERK kinases in neutrophils, 18 and only via ERKs in osteoblasts. 19 These results indicate that MK2 may act as a dual kinase downstream of p38 and ERK pathways by a tissue-specific mechanism. The opposite effects of MK2 in Ras-transformed mouse cells and human colon cancer propelled us next to explore if this distinct MK2 activity may result from its different activating pathways. Human kidney 293T epithelial cells were first used to assess endogenous MK2 phosphorylation in response to a mitogenic stimulus (phorbol ester [TPA]) or a stress agent arsenite (ARS). Results in Figure 3A showed that the level of phosphorylated MK2 (p-MK2) protein expression is increased after incubation of cells with both agents. Although TPA-induced p-MK2 is inhibited by neither a MEK inhibitor PD98059 (PD) nor a p38 inhibitor SB203580 (SB), ARS-induced p-MK2 is significantly suppressed by both inhibitors. Since TPA and ARS increase levels of p-ERK, p-p38α, and p-MK2 proteins, these results suggest that MK2 signals as a dual kinase downstream of ERK and p38α in human 293T cells. Similarly, both TPA and ARS also induce p38/ERK/MK2 phosphorylations in human colon cancer cells (Fig. 3B).
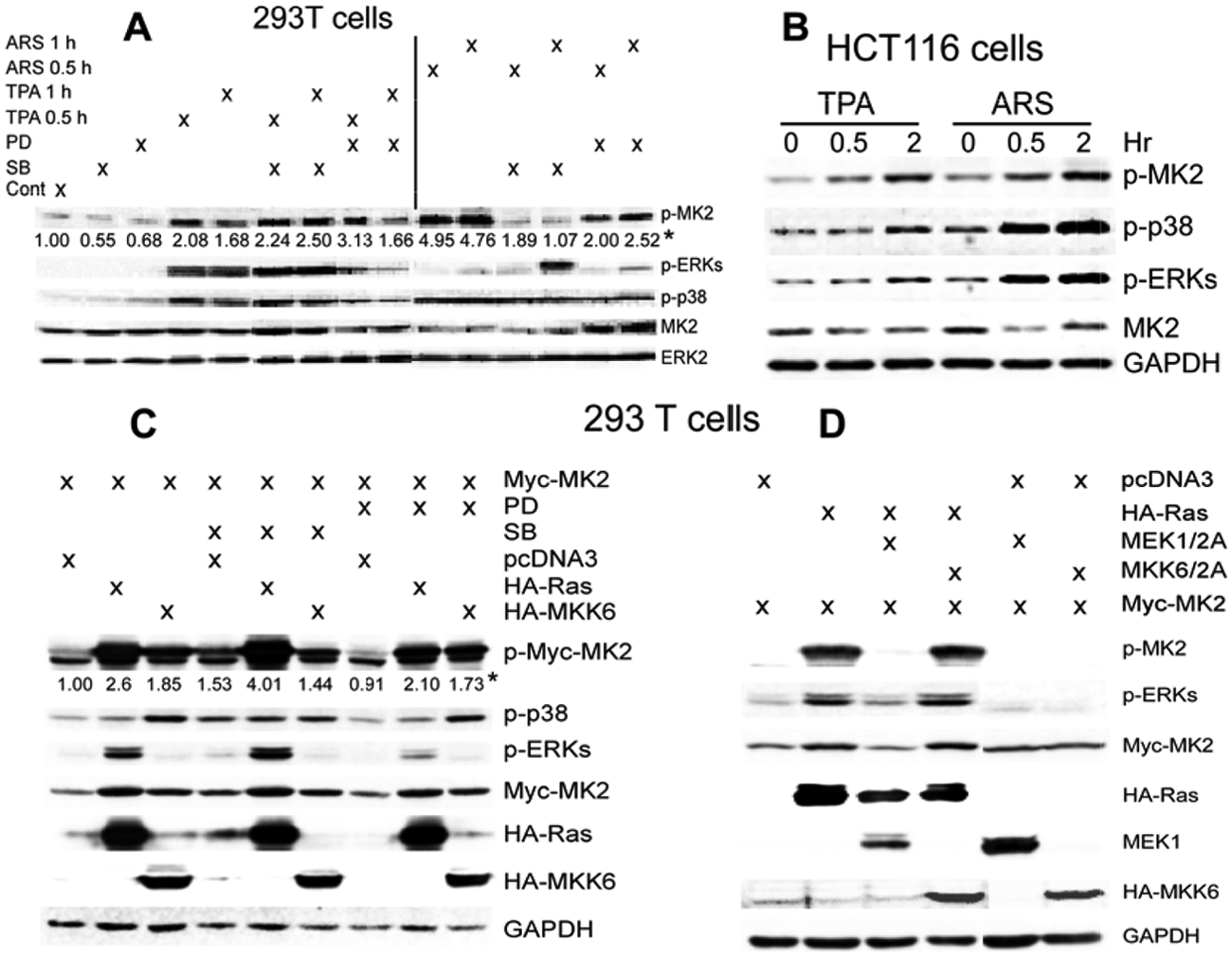
MK2 is activated by both mitogenic and stress stimuli downstream of ERK and p38 pathways in human 293T and colon cancer cells. (
To further confirm this observation by genetic means, 293T cells were transiently co-expressed with oncogenic H-Ras or constitutively active MKK6 together with WT MK2, and their regulatory effects on MK2 phosphorylation with and without PD or SB preincubation were examined. Results in Figure 3C again showed that both Ras and MKK6 strongly increase p-MK2 levels, which generally couples with their stimulatory effects on expression levels of the exogenous Myc-MK2. Of interest, Ras-induced p-MK2 was enhanced by SB but suppressed by PD, whereas MKK6-resultant p-MK2 was inhibited by both agents, albeit less substantially, thus further indicating the dual kinase property of MK2. Consistent with this notion, Ras-induced p-MK2 is blocked by co-expression of dominant-negative MEK1 (MEK1/2A) but not MKK6 (MKK6/2A) (Fig. 3D), indicating a role of ERK in Ras-stimulating MK2 as we previously reported. 5 These results together suggest that MK2 is a dual kinase downstream of ERK and p38 MAPKs in human cells, and the required role of ERK in Ras-induced MK2 phosphorylation suggests its potential involvement in MK2 oncogenic activity.
MK2 is activated only by stress but not mitogenic signals in mouse cells via a p38α-dependent mechanism
To assess whether MK2 is differently activated by mitogenic and stress signaling downstream of p38α in mouse cells, p38α+/+ and p38α−/− MEFs were pulse-treated with TPA and ARS, and MK2 phosphorylation was then examined by Western blotting. Results in Figure 4A showed that TPA and ARS both increase ERK phosphorylation, although p38α is predominantly phosphorylated by treatment with ARS in p38α+/+ cells. A faint p-p38 band after ARS in p38α−/− cells may be from another p38 family protein as previously observed. 20 The lack of p-MK2 in p38α−/− cells under these conditions is consistent with the previous observation that MK2 is a direct substrate for p38α 21 and supports the notion that MK2 is a stress kinase downstream of p38α. Endogenous MK2 protein expression is also decreased in p38α−/− cells as recently reported. 22 Of interest, treatment with TPA increases ERK but not MK2 phosphorylation in both WT and KO p38α (albeit less) MEFs as opposed to those observed in human colon cancer cells. Since ARS-induced p-MK2 is abolished in p38α−/− cells and TPA-resultant p-ERK uncouples with MK2 activation in both p38α WT and KO MEFs, these results suggest that MK2 may only act as a stress kinase downstream of p38α but not ERK in these mouse experimental tumor cells.
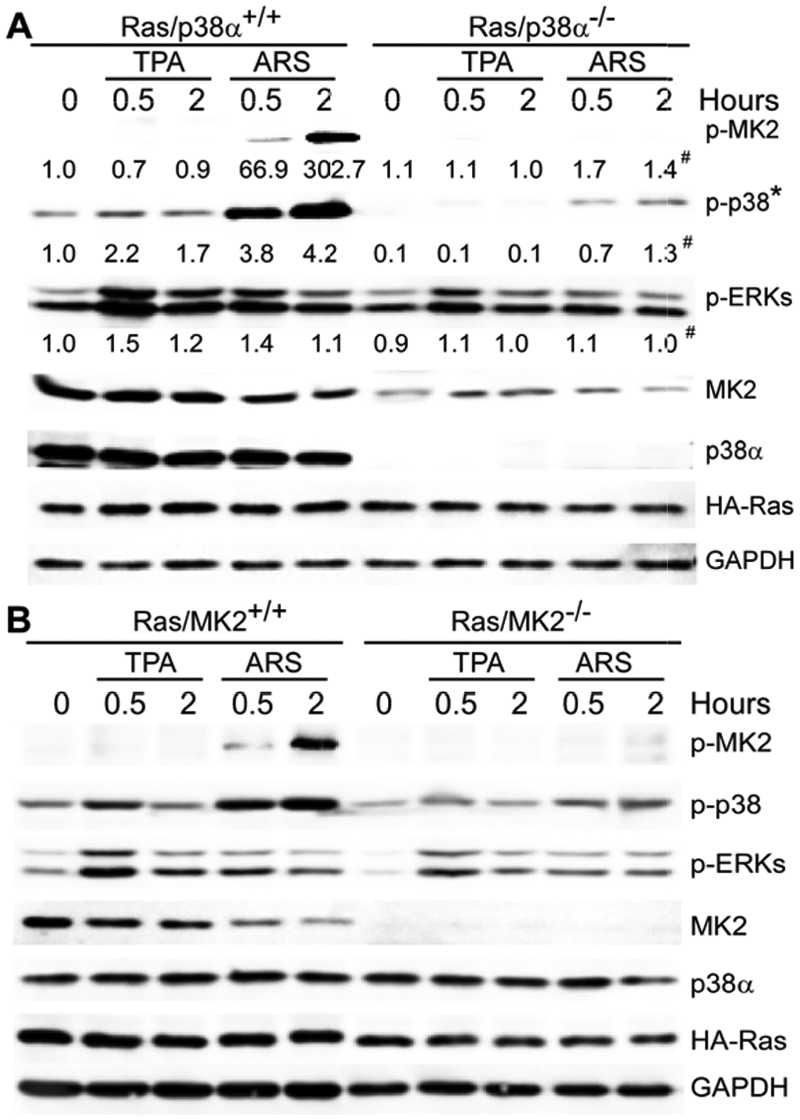
TPA activates ERK but not MK2, while ARS requires p38α to induce MK2 phosphorylation in mouse Ras-transformed cells. (
To further confirm this observation, the same analysis was performed in Ras-transformed MK2+/+ and MK2−/− MEFs. Results in Figure 4B showed that again, these reagents increase levels of p-ERK as well as p-p38α proteins and that MK2 is phosphorylated only after treatment with ARS but not with TPA. Decreased p38α phosphorylation in MK2−/− cells in response to both agents may be due to the lack of a MK2 stabilization effect as previously reported. 23 These results together indicate that in contrast to MK2 signaling through both p38α and ERK pathways in human 293T and colon cancer cells, MK2 is only activated by p38α-dependent stress but not by ERK-associated mitogenic signaling in mouse cells.
MK2 decreases cellular levels of ROS in experimental mouse tumor cells but increases its production in human colon cancer
ROS are molecules or ions formed by the incomplete 1-electron reduction of oxygen that can exhibit either cancer-promoting or cancer-suppressing activity. 24 Previous studies have shown that ROS signaling is involved in the transformation process by both oncogenic H-Ras 25 and K-Ras. 26 Moreover, a recent study showed that p38α inhibits Ras-induced ROS accumulation and that increased ROS signaling is coupling with decreased p38α phosphorylation and enhanced tumorigenicity. 25 Direct ROS regulatory effects of the p38α pathway in human cancer, however, have not been demonstrated. To investigate if the distinct Ras regulatory effects of MK2 in MEFs and human colon cancer cells couple with its different ROS regulations, intracellular ROS was measured by a fluorescence microscopy and plate reader. In the absence of Ras oncogene expression, levels of cellular ROS are undetectable, which, however, are substantially increased in MK2 KO but not WT MEFs following Ras transformation (Fig. 5A), indicating that MK2 inhibits Ras-induced ROS production in these cells. This conclusion is further supported by a decrease in ROS accumulation by the MK2 rescue in these cells (Fig. 5B). Since ROS is required for Ras transformation 26 and MK2 inhibits Ras tumorigenesis in MEFs, MK2 may inhibit Ras oncogenesis by suppressing ROS production.
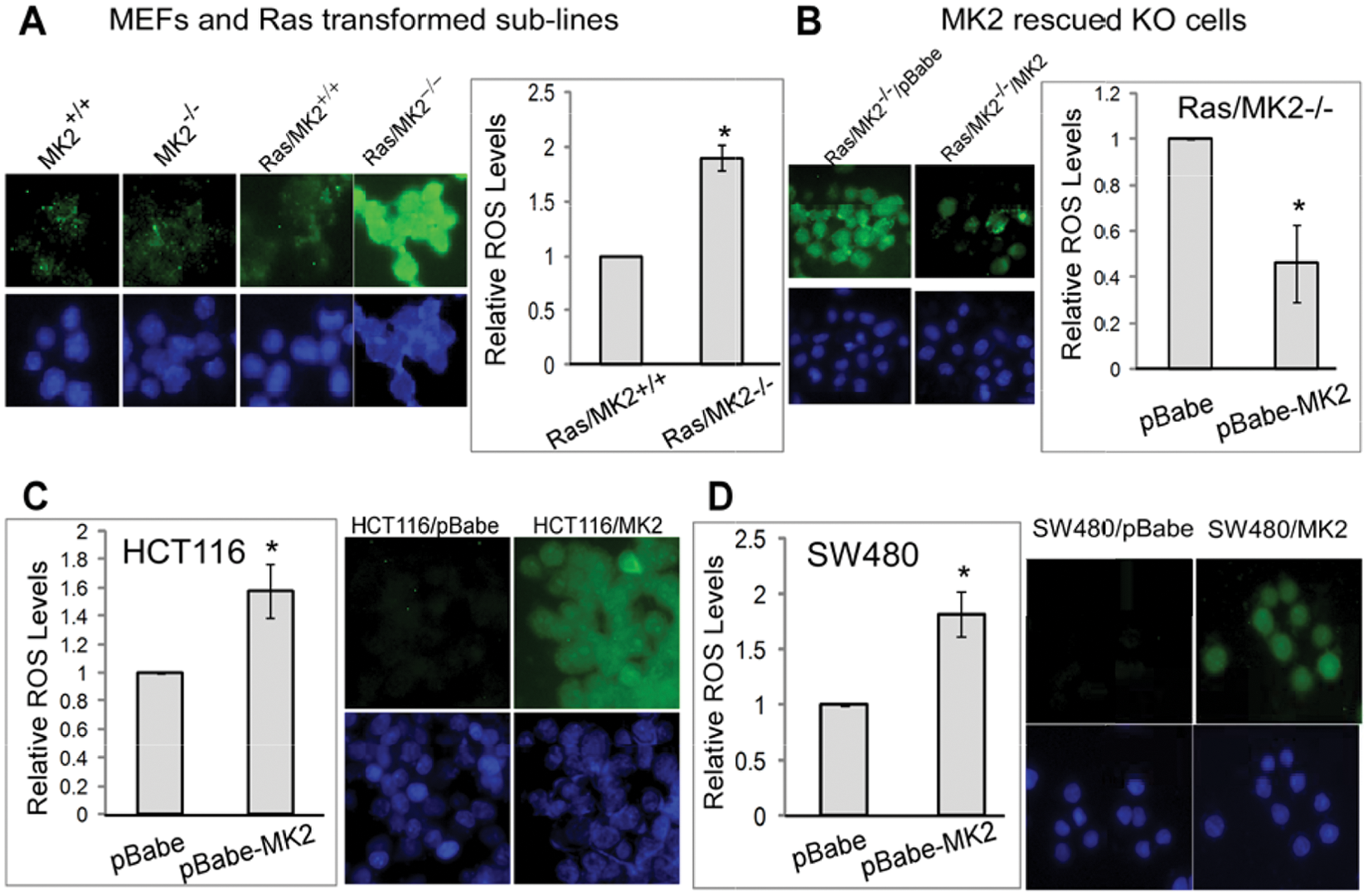
MK2 decreases cellular ROS levels in transformed mouse cells but increases its accumulations in human colon cancer cells. (
Human colon cancer cells were next analyzed for MK2 regulatory effects on ROS production. Since endogenous ROS in the absence of stress in these cells is undetectable, we focused on analyzing effects of MK2 activation. In this regard, HCT116 and SW480 cells stably transfected with a constitutively active MK2 were assessed for ROS accumulation. Surprisingly, in contrast to MEFs, the MK2-forced expression in both cell lines significantly increases ROS production (Fig. 5C and 5D). These results together reveal an opposing activity of MK2 in regulating ROS production in mouse experimental and human colon cancer cells.
ROS is required for the malignant growth in both transformed mouse and human colon cancer cells
The required role of ROS in Ras transformation 26 as well as the coupling of MK2 regulating Ras-dependent growth with ROS production suggests that MK2 may regulate Ras oncogenesis by altering ROS production. To directly test this hypothesis, HCT116 human colon cancer and Ras-transformed WT and KO MK2 cells were incubated with antioxidants glutathione (GSH) and N-acetylcysteine (NAC) and examined for soft agar growth and protein expression. Incubation of HCT116 cells with both ROS inhibitors significantly suppresses the colony formation (Fig. 6A, left), indicating a required role of ROS in human colon cancer growth. Since MK2 increases ROS accumulation in these cells (Fig. 5C), these results indicate that MK2 may increase the colon cancer growth by increasing cellular ROS production. Interestingly, the ROS inhibition by both agents also decreases the colony formation in Ras-transformed MEFs (Fig. 6A, right). Because MK2 decreases the ROS production and suppresses the colony/tumor growth in these transformed mouse cells (Figs. 1 and 5A and 5B), these results suggest that MK2 inhibits the malignant growth by decreasing ROS production. Western analyses showed that under these conditions, both GSH and NAC failed to show substantial effects on p-p38, p-ERK, and p-MK2 proteins, indicating that ROS may act downstream of ERK/p38/MK2 to promote the malignant growth (Fig. 6B and 6D).
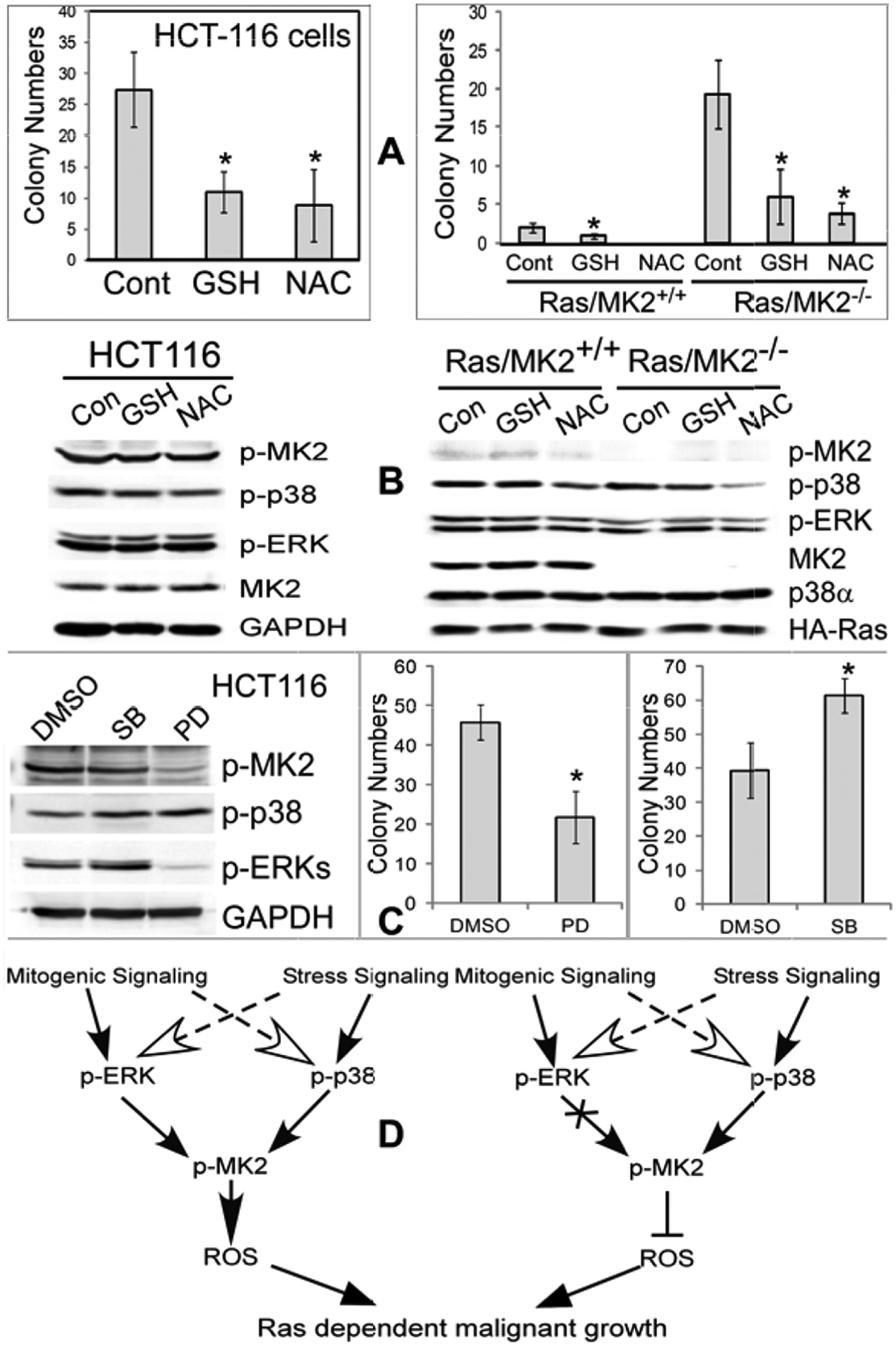
The role of ROS in the malignant growth of transformed mouse cells and human colon cancer cells downstream of MK2. (
The endogenous ERK but not p38 activity is required for intrinsic MK2 phosphorylation and malignant growth in human colon cancer cells
Comparative analysis has shown that MK2 signals downstream of p38α but not ERKs in Ras-transformed MEFs but acts as a p38/ERK-activated dual kinase in human cells. Experiments with both MK2 KO/depletion and expression further showed that MK2 inhibits Ras oncogenesis in MEFs but increases the growth in human colon cancer cells. These results together indicate that MK2 may propagate mitogenic signal components of ERK pathways to increase the malignant growth, which is available in human but absent in mouse cells. If this is the case, ERK, but not p38, inhibition would block both MK2 phosphorylation and human cancer growth. To directly test this possibility, HCT116 cells were incubated with and without the MEK inhibitor PD or the p38 inhibitor SB for 24 hours and analyzed for colony growth and protein expression. Results in Figure 6C showed that PD inhibits both ERK and MK2 phosphorylation, which couples with its suppressive effect on the colony formation, while SB increases the growth. The growth-increasing effect of SB is associated with its moderate activation of p-ERK, which may result from its Raf-1 stimulatory role in the cellular system as previously reported. 27 Although PD may act through additional cellular targets to regulate growth, decreased MK2 phosphorylation by the ERK inhibition that couples with reduced malignant growth strongly suggests that MK2 may signal downstream of ERK to promote human cancer growth (Fig. 6C and 6D).
Discussion
Studies from several laboratories have shown a suppressor role of the p38α pathway in Ras tumorigenesis in experimental cancers in vitro and in vivo.5,6,8,25,28 Moreover, knocking out p38α 29 and its substrate PRAK in mice 8 was further shown to increase skin or liver tumor development in response to chemical carcinogens, suggesting that the p38α/PRAK pathway may act as a general tumor suppressor in experimental cancer. Our studies presented here for the first time show that MK2, a direct p38α substrate, inhibits Ras oncogenesis in the mouse system but promotes the growth of Ras-dependent and -independent human colon cancer. Moreover, the opposite MK2 activity was found to directly link to its distinct ROS regulatory effects that couples with its different activations by stress and mitogenic signaling through p38 and ERK pathways. A demonstration of distinct activities of MK2 in regulating mouse transformed experimental and human cancer growth will contribute significantly to our understanding of regulating the malignant development through controlling MAPK signaling.
The oncogenic activity of MK2 in human colon cancer may be due to its dual kinase property downstream of p38 and ERK pathways in which MK2 may convert the mitogenic ERK signaling component into a growth response. This conclusion is suggested by the fact that in human colon cancer cells, MK2 is activated by both mitogenic and stress stimuli downstream of p38 and ERK pathways, ERK inhibition suppresses the colony formation as well as MK2 phosphorylation, and MK2 depletion inhibits and MK2 transfection increases the malignant growth. While substantial studies have revealed a critical role of MK2 in stress response,30,31 our results may be the first showing that the regulation of MK2 expression alone is sufficient to control human cancer growth. The oncogenic effect of MK2 in human cells is consistent with the previous observation in which MK2 is required for vascular endothelial growth factor (VEGF)–induced human cell migration 16 and for transforming growth factor β (TGFβ)–resultant human prostate cancer cell invasion. 32 The required role of the ERK activity in mitogenic signaling stimulation of MK2, on the other hand, is consistent with the previous observation that the ERK pathway participates in attenuating the p38-induced G2 delay of the cell cycle. 33 In agreement with the oncogenic activity and the ROS stimulatory effect of MK2, recent studies also showed the survival effect of MK2 activation in stress response through activation of the Akt/Hsp27 axis in rat PC6 cells and Drosophila.34,35 These results, together with a recent finding of increased p-MK2 expression in clinical human thyroid cancers, 36 indicate that MK2 may function downstream of the ERK pathway to promote human cancer growth and/or progression. Additional experiments are needed to demonstrate if MK2 is required for Ras-dependent and -independent human colon cancer growth in vivo and whether higher levels of p- MK2 proteins are correlated with increased p-ERK in primary colon cancer tissues.
The oncogenic activity of MK2 may be derived from its capacity to stimulate ROS production (Fig. 6C). This conclusion is supported by our experimental evidence showing that MK2 increases the ROS production and the malignant growth in human cancer cell lines but inhibits both in mouse experimental tumor cells, and ROS is required for the malignant growth in both mouse and human systems. Mechanisms for the opposing ROS regulatory effects by MK2 in 2 systems, however, are not understood at this time but may relate to different MK2 activation pathways. This assumption is indicated by the fact that p-MK2 is induced by both TPA and ARS via the ERK and p38α pathways in humans but only responsive to ARS (but not TPA) in MEFs despite the ERK activation. MK2 was previously shown to receive signaling from ERK and p38 pathways in human neutrophils, 18 whereas ERK and MK2 can collaborate to promote colon cancer survival as demonstrated by the fuzzy logic modeling analysis. 37 Although studies from Cai et al. group recently showed that MK2 may signal downstream of p38 to increase cell survival in response to ROS exposure, 34 our results may be the first to show that MK2 inhibits and stimulates ROS production in mouse and human cells, respectively. These results together indicate a theory that MK2 may function as a dual kinase downstream of ERK/p38α pathways in humans but primarily as a p38α-activated stress kinase in the mouse system, which may lead to its opposing regulatory effects on ROS production and on malignant growth. It remains, however, to determine whether a specific regulation of the ERK activity alone will only alter cellular ROS in human but not mouse cells and if an artificial ROS elevation promotes the malignant growth both in mouse experimental tumors and in human cancers.
It is well established that different signaling components are required to transform murine and human cells. 38 While the growth inhibition of Ras-transformed mouse cells and the stimulation of Ras-dependent and -independent human colon cancer cells by MK2 may indicate its species-specific effect (Fig. 6D), several additional factors must be considered. For example, MK2 may regulate Ras activity by cell/tissue- and/or Ras isoform–specific mechanisms, and analyses of a different mouse cell line with another type of Ras-dependent human cancer may yield a different result. The growth enhancement effect of MK2 on both K-Ras–mutated and –nonmutated human colon cancer cells, on the other hand, would argue against the possible involvement of Ras isoforms in these regulations. In addition, MK2 may have distinct regulatory activities in tumor initiation and in tumor maintenance. An ideal model would include studying MK2 regulatory effects in mouse versus human colon cancer with both containing endogenous activated K-Ras– or H-Ras–transformed mouse versus human fibroblasts. Therefore, whether MK2 regulates Ras oncogenesis by a species-specific mechanism remains to be established further. Nevertheless, ROS is required for both H-Ras and K-Ras oncogene activity,26,39 and MK2 activation by expressing the same cDNA leads to opposite effects on the malignant growth as well as on ROS production in both mouse and human systems. We may therefore still be able to conclude that MK2 regulates Ras oncogenesis through altering ROS production. Studies of MK2 stimulation of ROS production and of human colon cancer growth may reveal a novel therapeutic approach against human malignancies.
Materials and Methods
Reagents, expression constructs, and antibodies
ARS, TPA, NAC, and 2′,7′-dichlorofluorescin diacetate (DCF-DA) were purchased from Sigma (St. Louis, MO). p38 inhibitor SB203580 and ERK inhibitor PD98059 were obtained from Calbiochem (San Diego, CA). Reduced GSH was from Boehringer Mannheim (Mannheim, Germany). Retroviral pLZRS–HA–H-Ras (G12V) was previously described, 12 and pBABE-MK2 was a gift from Dr. S. Huang. 16 HA-tagged H-Ras (G12V), dominant-negative MEK1/2A, dominant-negative MKK6/2A, and constitutively active MKK6/2E were described previously.5,12 Mouse Myc-tagged MK2 was kindly provided by Dr. M. Gaestel. 21 To deplete endogenous MK2 expression in human colon cancer cells, the target sequence (5′-CCATCATCGATGACTACAA-3′) was cloned into the pSUPER retroviral vector via Bgl II and Hind III sites as previously described. 12 Phospho-p38 (p-p38), p-ERK, and MK2 antibodies were purchased from Cell Signaling Technology (Danvers, MA), whereas p-MK2, MEK1, MKK6, ERK2, p38α, and GAPDH were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies against FLAG (M2) and HA (clone 12CA5) were from Sigma, against c-Myc from Roche (Basel, Switzerland), and against pan-Ras from Oncogene Science (Cambridge, MA).
Cell culture, transfection, infection, gene expression, and gene knockdown
WT and KO MK2 immortalized MEFs were kindly provided by Dr. M. Gaestel,11,40 and WT and KO p38α MEFs were provided by Dr. A. Nebreda.20,25 Human HEK-293T cells (293T) and human colon cancer HCT116 and SW480 cells were purchased from ATCC (Manassas, VA). The K-Ras KO HCT116 subline HKE3 cells were a gift from Dr. S. Shirasawa. 14 Cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin and streptomycin at 37°C, 5% CO2.
For transient expression, plasmids were transfected by calcium phosphate. Cells were typically collected in 1X loading buffer for Western analyses 48 hours later. For virus production, plasmids were transfected into packaging cells, and resultant viral supernatants were collected for infecting target cells as previously described. 12 For the rescue experiment, MK2−/− cells were stably co-expressed with LZRS–H-Ras and pBABE-MK2. To establish stable Ras-transformed WT and KO p38α and MK2 cells, LZRS–HA-Ras retrovirus or the vector was introduced into these cells through the puromycin selection. To stably overexpress MK2 in human colon cancer cells, cells were infected with pBABE retrovirus expressing a constitutively active MK216 or a vector and selected with puromycin. To deplete endogenous MK2, siLuc and siMK2 were delivered into target cells by retroviral vector pSUPER, and stable silenced cell lines were obtained for analyses.
Colony formation assays and mouse experiments
Ras-transformed MK2+/+ and MK2−/− cells and MK2 stably expressed or depleted HCT116 and SW480 cells were plated at 1 × 104 cells per 60-mm dish in growth media containing 0.33% SeaPlaque agarose (Cambrex Bio Science Rockland, Rockland, ME). Colonies from an entire 60-mm plate were photographed and the numbers from 20 fields of each plate manually counted. 12 To examine effects of ROS inhibition on colony formation, cells were cultured with GSH for 3 days or NAC for 4 days both at 10 mM, which were then plated for colony formation assays with the top layer of soft agar containing the same concentration of inhibitors for about 2 weeks. Colony formation for Caco2 cells was performed as previously described. 41 Animal experiments were conducted in accordance with the approved protocols of the Medical College of Wisconsin Institutional Animal Care and Use Committee. Briefly, Ras/MK2+/+ and Ras/MK2−/− cells were collected from cell culture. Following washing with cold PBS, 2 × 106 cells in 0.1-mL volume were subcutaneously injected into athymic nude mouse (Harlan) at both front flanks, and the tumor volume (= π abc/6, where a, b, and c represent tumor length, width, and height, respectively) from this group of 5 mice was measured every 2 to 3 days as previously described. 42 The tumor was also removed, weighted, and photographed by the end of the experiment.
Chemical treatment, Western blotting, and ROS assay
To assess the effects of mitogenic and stress signaling on MK2 phosphorylation, TPA (200 ng/mL) and ARS (200 µM) were added into the culture for 30 minutes or 2 hours after a 24-hour preincubation with SB, PD, or solvent control. For Western blotting, cells were lysed in 1X loading buffer, and after heating, the lysates were separated by SDS-PAGE, which was transfected to a nitrocellulose membrane using a liquid transfer system (Bio-Rad, Hercules, CA). All of the following procedures were the same as those previously described. 12 To visualize ROS levels, proliferating cells were grown on cover slips at 3 × 105 in 2 mL on a 6-well plate. The next day, cells were washed once with warm PBS and incubated with 5 µM DCF-DA in warm PBS for 10 minutes at 37°C. 25 Following an additional incubation with complete medium, cells were fixed with 4% formalin for 15 minutes at room temperature, and cover slips were examined under a fluorescent microscope (Leica, Wetzlar, Germany). To measure intracellular ROS levels, cells were seeded onto a 24-well plate in triplicate at 1 × 105 in 0.5 mL of medium per well. The next day, cells were incubated with the DCF-DA solution and washed with PBS, and fluorescence signals were measured by a plate reader (CytoFluor, Applied Biosystems, Foster City, CA) (at excitation wavelength of 450 nm and emission wavelength of 530 nm). 43 Average absorbance of experimental groups was subtracted by that from background wells and expressed as a relative change over the individual control.
Footnotes
Acknowledgements
The authors thank Drs. M. Gaestel, A. Nebreda, J. Han, S. Huang, and S. Shirasawa for providing important reagents; Drs. M. Gaestel and A. Nebreda for critically reading the article; and Chen’s laboratory members for useful discussions.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from the National Institutes of Health (2R01 CA91576), Department of Veterans Affairs (Merit Review), and Wisconsin Breast Cancer Showhouse of the Medical College of Wisconsin Cancer Center (to G.C.).