
Abstract
Multiple growth factors and extracellular signals can lead to activation of the c-Jun amino N-terminal protein kinase (JNK) pathway. Activation of JNK can in turn lead to a multitude of downstream changes in phosphorylation and transcriptional activation within the cell. Mapping the upstream and downstream connectivity within the JNK network reveals an enrichment of bi-fan and feed-forward network motifs formed immediately upstream and downstream of JNK. In addition, negative feedback loops also exist through transcriptional activation of phosphatases that target the JNK pathway. The combinations of these motifs allow flexibility and tunability in signal integration and processing within the JNK network and may confer the wide range of biological responses that can be regulated by JNK activation. In this review, we highlight the pathways and motifs leading to JNK activation and its downstream signaling as well as the complexity in isoforms within this network.
Introduction
The c-Jun amino N-terminal protein kinases (JNKs) were initially identified in the early 1990s as the protein kinases that bind to a specific region of c-Jun and are involved in the phosphorylation of c-Jun at the N-terminal Ser-63 and Ser-73 sites in the transcriptional activation domain.1,2 This phosphorylation disrupts the c-Jun and JNK complex and the mutations responsible for this disruption effect Ha-Ras and ultraviolet (UV) responsiveness.1,3 Tumor activated Ras stimulates this phosphorylation3-5 and is also involved in the UV response leading to increased c-Jun activity, 6 thus indicating that the potential activation of JNK by UV response may play a major role in tumor growth. JNK protein kinase is thus identified to be activated by UV responsiveness and Ha-Ras mutations1-6 and has a major form of a 46-kD polypeptide. 1 The sites phosphorylated on c-Jun by JNK consist of a Ser residue followed by a Pro and an acidic residue. This identifies JNK as a member of the Pro-directed kinases that includes the mitogen-activated protein kinases (MAPK) family and the cyclin dependent protein kinases. The activation of MAPKs requires dual phosphorylation at Thr and Tyr within a conserved sequence located between kinase subdomains VII and VIII. 7 These Thr and Tyr residues are conserved in JNK1 and mutations at these residues blocked UV-induced phosphorylation and activation and are thus involved in the dual-phosphorylation site motif: Thr*-Pro-Tyr*.
Ten isoforms of JNK have been identified and are encoded by 3 distinct genes: JNK1, JNK2, and JNK3, the transcripts of which are spliced to yield 4 isoforms of JNK1, 4 isoforms of JNK2, and 2 isoforms of JNK3. Of these, JNK1 and JNK2 are found everywhere whereas JNK3 is expressed mainly in the heart, testes, and brain. 8 JNK isoforms are activated in rheumatoid arthritis (RA) synoviocytes and synovium and play important roles in the production of cytokines and extracellular matrix regulation by enhancing production of metalloproteinase.9,10
JNK Pathways and Networks
The stress activated protein kinases (SAPK)/c-Jun amino N-terminal kinases (JNK) are members of the MAPK family and are activated by environmental stress and cytokines. SAPK/JNK translocates to the nucleus where it can regulate the activity of multiple transcription factors. Stress signals are delivered to this cascade by small GT Pases of the Rho family (Rac, cdc42, Rho). JNK can phosphorylate transcription factors, including c-Jun, and promote activator protein-1 (AP-1) mediated transcription. 11 JNKs are activated by MAP 2 Kinases MKK-4, MKK-3, MKK-6, and MKK-7. These are in turn activated by the MAP 3 kinases: MEKK, mixed lineage kinases MLK-2, MLK-3,12,13 transforming growth factor-β-activated kinase-1 (TAK-1), 14 tumor progression locus-2 (TPL-2), 15 and apoptosis signal-regulating kinase-1 (ASK1). 16
Activated forms of Rac and Cdc42Hs are known to activate the c-Jun kinase JNK/SAPK. 17 The stress signals are delivered to this cascade via the small GTPases of the Rho family (Rac, Cdc42, Rho). Rho, Rac, and Cdc42 control the signal transduction pathways that are essential for cell growth. JNK activation was also observed in response to Dbl, an oncoprotein that acts as a nucleotide exchange factor for Cdc42Hs. 18 JNKK, a dual specificity kinase that functions between MEKK and JNK, was identified to activate JNK. JNKK activated JNK and MAPK and p38 but not ERKs. 19 Specific ERK activators, MEK1 and MEK2, are activated by Raf-1 but the JNK activator MKK4 is not a substrate for Raf-1, implying that Raf-1 does not affect JNK activation. 19
JNK activation is mediated by phosphorylation on both Thr and Tyr residues by dual specificity MAPK kinase (MAPKK). MAPK kinase 7 (MKK7) and MAPK kinase 4 (MKK4) have been identified as specific activator of the JNK signaling. 20 MEKK4, also known as the stress activated protein kinase/ERK kinase or JNK kinase, is known to activate both p38 and JNK pathways. 21 Members of the MEKK family of serine/threonine kinases were the first MAPK kinase kinase shown to activate JNK. MEKK1, MEKK2, and MEKK4 can activate JNK. Upstream MAPK kinase and MAPK kinase kinase-5 (MAPKKK5) are also known to activate the JNK cascade by directly phosphorylating MKK4. MEKK3 has also been known to strongly induce JNK activity in cells 22 and may mediate JNK activation. In the study by Deacon and Blank, 21 the authors have shown that MKK6 and MKK7 are specific for activation of p38 and JNK1. The MEKK isozymes can function as MAP 3 kinases in the ERK and JNK signaling pathways. It is also observed in the above that MEKK4 has greatest selectivity for MKK4/JNK cascade and does not activate ERK or MEK. MEKK3 represents a MAP 3 kinase that activates MKK7 in the JNK pathway. MEKK1 and MLK2 have also been shown to activate MKK7. 23 The JNK-interacting protein, JIP, is a molecular scaffold consistent with substrate selectivity of MEKK3 for MKK7 and is capable of binding JNK, MKK7, and MEKK3 and activate JNK signaling via this cascade. 24
MAPK signaling pathways, which were originally called the extracellular signal-regulated kinase (ERK), are known to be activated by cellular stress and in response to extracellular stimuli. MAPK family includes the ERKs, JNKs, and the p38 kinases. Of these, JNK and p38 are activated by cellular stress. These stress activated pathways are known to be involved in apoptosis and tumor growth in various cell types. 11 On activation by the upstream MAP 2 kinases, the phosphorylated JNK translocates to the nucleus where it further phosphorylates and transactivates c-Jun and regulates the activity of multiple transcription factors.
JNKs once translocated to the nucleus can phosphorylate multiple transcription factors including JunD, ATF2, ATF3, Elk-1, Elk-3, p53, RXRα, RARα, AR, NFAT4, HSF-1, and c-Myc. 25 In the context of apoptosis, the nuclear activity of JNK potentially leads to an increased expression of pro-apoptotic genes and/or a decrease in the expression of pro-survival genes. 11
The interacting components of JNK activation and its effector downstream pathways present a complex network structure of interlinking network motif structures. These motifs include bi-fans as well as feed-forward loops. 26 These basic network motifs that are overrepresented in biological systems 27 can confer dynamic tunability and flexibility in signal processing through the JNK activation network. Several groups have modeled the dynamic properties of these network structures and focused specifically on JNK activation in response to a variety of ligands and activation conditions. More about this can be found in various studies.28-31
Functional Roles of JNK Activated Networks
The MAP kinase pathway is involved in cell proliferation, differentiation, development, inflammatory response, and apoptosis. 32 Some of the new negative regulatory components of the JNK signaling pathway include the dual specificity MAPK kinase phosphatase MKP7, which inactivates some JNK isoforms. 33 The heat shock protein Hsp72 was reported to inhibit JNK. 34 Nitric oxide was reported to inhibit JNK by causing the covalent modification of the enzyme by S-nitrosylation. 35 The prolyl isomerase Pin1, which interacts with some phospho-Ser/Thr-Pro sites, is reported as a positive regulator of the JNK signaling pathway. 36 Pin1 also bind to the c-Jun phosphorylated by JNK on Ser-63 and Ser-73.
JNK signaling also contributes to apoptotic signaling through the phosphorylation of pro-apoptotic proteins. 11 JNK can phosphorylate Bcl-2, which results in the migration of Bcl-2 from the mitochondrial membrane and dissipation of the mitochondrial membrane potential. 37 At the outer mitochondrial membrane, JNK can interact and phosphorylate a scaffold, Sab (SH3BP5). 38 This interaction of Sab with JNK occurs through a mitogen-activated protein kinase interacting motif (KIM). JNKs are also responsive to mitochondrial signals. TNF-α induced reactive oxygen species (ROS), which are by-products of cellular respiration and whose accumulation is suppressed by mitochondrial superoxide dismutase, cause oxidation and inhibition of the JNK. 39 This results in sustained JNK activation, which is required for necrotic cell death. The increase in ROS levels in the mitochondria causes damage to the reactive cysteines in phosphatases and metabolic enzymes. 40 JNK is activated by ROS through upstream kinases, primarily apoptosis signaling regulated kinase (ASK1). Anisomycin activates JNK signaling and stimulates JNK-dependent ROS production. 40 JNK signaling mediates amplification of ROS production during stress.
MAP kinase are expressed and activated in the RA synovium and have been the focus of the drug developmental programs due to their prominent role in control of cytokines, chemokines, degradative enzymes, programmed cell death, and cell proliferation. JNK1 but not JNK2 is shown to be critical for joint swelling and destruction in a serum transfer model of arthritis. Guma et al. 41 have also observed that a JNK-inhibiting peptide abrogated joint swelling in established disease, suggesting that JNK is also involved in the progression of arthritis.
Activated nuclear JNK1 binds to specific sites in the genome, a subset of which is estrogen regulated. At the estrogen induced sites, estrogen receptor-α (ERα) is required for binding of the JNK1. At estrogen regulated promoters, JNK1 functions as a transcriptional co-regulator of ERα required for efficient estrogen dependent transcription of these genes and for downstream cell growth response. The expression of ERα in cells is a well-known prognostic indicator for breast cancer. JNK1 is regulated by the phosphorylation of a Thr-Pro-Tyr motif by upstream MAPK kinases. Recent studies 42 have shown that JNK1 is required for full estrogen-dependent growth of breast cancer cells, and thus estrogen and JNK1 signaling pathways collaborate to control the proliferation in breast cancer cells.
A recent study has shown that a mammalian MAP kinase, JNK, is recruited to a large set of gene promoters in response to differentiation signals and that it facilitates the activation of target genes by directly modifying chromatin components. This represents a new pathway by which MAPK regulates gene expression and expands the number of target genes for downstream of JNK activation. 43 Tiwari et al. 43 have also found that JNK target promoters are preferentially enriched for phosphorylated H3S10. JNK phosphorylates H3S10 in vitro, and inhibition of JNK phosphorylation reduces phosphorylation at this site and diminishes target gene expression in neuronal cells. It was also observed that during ES cell differentiation, the mRNA levels of MAPK-9 (encoded JNK2) stay constant, whereas those of MAPK-8 (encoded JNK1) and MAPK-10 (encoded JNK3) are elevated in terminally differentiated pyramidal glutamatergic neurons (TNs).
c-Jun has emerged as a promising therapeutic target in clinical cancer treatment. 9 In tumor cells, the c-Jun protein expression level and activity are increased. The c-Jun gene is an early response proto-oncogene, and its protein product c-Jun is a major component of the AP-1 transcription factor complex. The most well-characterized phosphorylation sites of c-Jun are Ser63 and Ser73 located near the NH2-terminal. In the study by Li et al., 44 it is shown that knockdown of P21 activated protein kinase (PAK2) had no effect on either phosphorylation site and had no effect on phosphorylation of total c-Jun, but knockdown of PAK2 blocked AP-1 activity. PAK2 binds and phosphorylates c-Jun and PAK2 phosphorylates c-Jun at 5 threonine sites: Thr2, Thr8, Thr89, Thr93, and Thr286. Thus, PAK2 might play a role in suppressing AP-1 activity through modification of other c-Jun phosphorylation sites.
Very recently, it has been shown that the JNK/c-Jun signaling axis contributes to the transactive response DNA-binding protein-43 (TDP-43) induced cell death. 45 The overexpression of TDP-43 results in the increase of c-Jun N-terminal kinase phosphorylation, and TDP-43-induced motor neuronal cell death is attenuated by the co-incubation with a JNK inhibitor. It was reported by Suzuki and Matsuoka 45 that while the phosphorylation levels of JNK increases, the total JNK level is not increased by the TDP-43 expression.
The JNK signaling pathway plays a major role in stimulating programmed cell death (or apoptosis) through mitochondrial as well as transcriptional based mechanisms. Apoptosis plays a fundamental role during development of multicellular organisms, serving to sculpt tissues by removing excess cells, establish appropriate immune cell complements through the elimination of self-reactive cells, and maintain tissue homeostasis through regulation of normal cell turnover. 46 JNK has been suggested to be pro-apoptosis, anti-apoptosis, or have no role in apoptosis depending on the cell type and stimulus used. JNK can phosphorylate many cytoplasmic proteins, including Bcl-2 and Bax, which play a direct role in regulating the apoptotic response. JNK is required for IL-3 mediated cell survival through phosphorylation and inactivation of the pro-apoptotic Bcl-2 family protein BAD.47,48 JNK signaling cascade initiates apoptotic response to genotoxic stress. The DNA binding domain of p53 is capable of preventing MKP-5 mediated dephosphorylation of activated JNK whereby p53 binding to JNK regulates the level of kinase activity with stress-dependent stimulation of p53 levels resulting in sustained activation of JNK and induction of apoptosis. 49 P53 is capable of binding to a number of different cellular substrates and may act to recruit activated JNK to specific promoters where it might stimulate transcription through the selective activation of Fos-c-Jun heterodimers (AP-1) bound at specific promoter sites.
Conclusion
Activation of JNK can be accomplished through several receptors signaling pathways upstream from JNK and at the same time JNK can also activate several downstream effector pathways. Examining the core network motifs present in JNK signaling shows the presence of bi-fan motifs that are stacked on each other while at the same time containing numerous feed-forward loops. Figure 1 highlights some of these features. These complex network structures might play a large role in the multitude of biological responses that are triggered by JNK activation. In addition, these network motifs that are present upstream of JNK allow signals to be integrated and processed, thereby allowing flexibility and tunability in JNK signaling. Much more detailed systems analysis of the JNK network needs to be done incorporating both the dynamic properties as well motif features of the JNK network to fully understand the role of JNK in regulating diverse biological processes.
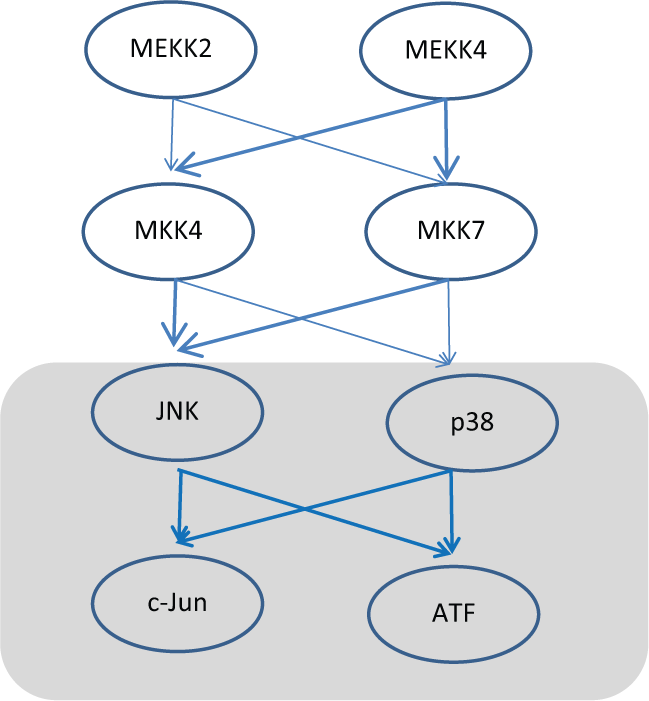
An example of the bi-fan between the 2 MAP-kinases, p38 and JNK, that phosphorylates and activates the transcription factors ATF and c-Jun. Highlighted in bold arrows are the feed-forward networks herein leading to JNK activation and the box highlights the coherent bi-fan motif between JNK/p38–ATF/c-Jun. MEKK2 = mitogen activated protein kinase kinase kinase 2; MEKK7 = mitogen activated protein kinase kinase kinase 7; MKK4 = mitogen activated protein kinase kinase 4; MKK7 = mitogen activated protein kinase kinase 7; JNK = c-Jun N-terminal kinase; P38 = mitogen activated protein kinase; c-Jun = Jun proto-oncogene; ATF = activating transcription factor.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: VS is supported by the Computational Cancer Biology Training Program, Cancer Prevention and Research Institute of Texas (CPRIT Grant No: RP101489).