
Abstract
NKX3.1 is a tumor suppressor down-regulated in early prostate cancers. A SNP (rs2228013), which represents a polymorphic NKX3.1(C154T) coding for a variant protein NKX3.1(R52C), is present in 10% of the population and is related to prostatic enlargement and prostate cancer. We investigated rs2228013 in prostate cancer risk for 937 prostate cancer cases and 1,086 age-matched controls from a nested case-control study within the prospective Physicians’ Health Study (PHS) and among 798 cases and 527 controls retrospectively collected in the Risk Factors for Prostate Cancer Study of the Victoria Cancer Council (RFPCS). We also investigated the interaction between serum IGF-I levels and NKX3.1 genotype in the populations from PHS and RFPCS. In the PHS, we found no overall association between the variant T allele in rs2228013 in NKX3.1 and prostate cancer risk (odd ratio = 1.25; 95% confidence interval = 0.92-1.71). A subgroup analysis for cases diagnosed before age 70 showed an increased risk (relative risk = 1.55; 95% confidence interval = 1.04-2.31) of overall prostate cancer. In this age-group, the risk of metastatic cancer at diagnosis or of fatal cancer was even higher in carriers of the T allele (relative risk = 2.15; 95% confidence interval = 1.00-4.63). These associations were not replicated in the RFPCS. Serum IGF-I levels were found to be a risk factor for prostate cancer in both study populations. The wild type NKX3.1 protein can induce IGFBP-3 expression in vitro. We report that variant NKX3.1 cannot induce IGFBP-3 expression, but the NKX3.1 genotype does not modify the association between serum IGF-I levels and prostate cancer risk.
Introduction
Adenocarcinoma of the prostate, like many malignancies, initiates in epithelial cells that acquire the precursor or gatekeeper mutations required for development of the malignant phenotype. In the majority of early prostate cancers a region of 8p21.2 is lost resulting in loss of the homeobox gene NKX3.1 that is expressed specifically in prostate luminal epithelial cells.1,2 Somatic inactivation of NKX3.1 in prostate cancer is reflected in decreased protein expression that is at first partial and then nearly complete at the time prostate cancer progresses to hormone-independence and metastatic disease.2,3
Loss of NKX3.1 expression is a very early event in prostate carcinogenesis. Gene targeting studies in mice showed that Nkx3.1 haploinsufficiency alone can predispose to prostate epithelial dysplasia and can cooperate with other oncogenic mutations to augment prostate carcinogenesis.4,5 Heterozygous Nkx3.1+/− mice have approximately two thirds the Nkx3.1 protein levels of wild type mice. A similar reduction in NKX3.1 protein levels is seen in human prostatic intraepithelial neoplasia and in primary human prostate cancer. 3 Not only is NKX3.1 down-regulated in preinvasive prostate cancer but also NKX3.1 expression is reduced in regions of inflammatory atrophy that are precursors for malignant transformation. 6 Inflammatory cytokines in these lesions can induce ubiquitination of NKX3.1 that targets the protein for degradation in the proteasome. 7 Thus, decreases in NKX3.1 can both predispose to and accompany prostate malignant transformation.
Although no somatic mutations of NKX3.1 have been found in prostate cancer, 8 there are 2 well-characterized genetic variants associated with prostate cancer. A missense mutation was found that altered the N-terminal cap amino acid of the third, DNA-binding, helix in the homeodomain from a threonine to an alanine NKX3.1(T164A) that conferred risk for early prostate cancer in a family. 9 A (rs2228013) that represents a polymorphic NKX3.1(C154T) coding for a variant protein NKX3.1(R52C) is present in 10% of the population and is related to prostatic enlargement and prostate cancer. 20
Growth suppression by NKX3.1 is affected, in part, by inducing expression of insulin-like growth factor binding protein-3 (IGFBP-3), a known growth suppressor protein and down-regulator of insulin-like growth factor-I (IGF-I) activity. IGF-I is a peptide growth factor that regulates cell growth, differentiation, and apoptosis by binding to the IGF receptor-I (IGFR-I). 10 IGFs are present in abundance in the circulation. Circulating IGF-I is bound mainly to IGFBP-3, one of the most abundant serum proteins. 11 Although IGFBP-3 can inhibit the interaction of IGF-I with its receptor at the cellular level, serum IGFBP-3 also serves to stabilize circulating IGF-I. 12 Because of the effects of IGF-I on cell growth, survival, and apoptosis, the influence of both serum IGF-I and IGFBP-3 concentrations on cancer risk has been studied by a number of investigators. 12 Serum IGF-I levels are associated with an elevated risk of prostate cancer in a variety of studies13-17 that have been confirmed by meta analyses,18,19 Here we show that NKX3.1(R52C) and a protein engineered for loss of the serine 48 phosphorylation site NKX3.1(S48A) do not activate expression of IGFBP-3. Consistent with this loss of function, we hypothesize that the presence of the variant NKX3.1 protein may interact with circulating serum IGF-I to affect prostate cancer risk. Determination of NKX3.1 genotype in 2 populations and analysis of serum IGF-I in the same study subjects is shown.
Results
Effect of NKX3.1(R52C) on IGFBP-3 expression and IGF-IR activation
Amino acid 52 affected by rs2228013 is an arginine, located in a consensus motif that is a site for phosphorylation at serine 48. Replacement of arginine 52 with cysteine decreases phosphorylation at serine 48 by 70%. 20 Thus, a missense mutation at serine 48 potentially generates a protein with analogous, but more absolute, loss of serine 48 phosphorylation compared to NXK3.1(R52C). Expression of NKX3.1(R52C) in PC-3 cells induced substantially less IGFBP-3 mRNA than did wild type NKX3.1 (Fig. 1A). The mutant NKX3.1(S48A) protein was also attenuated in IGFBP-3 induction, perhaps to a greater degree than NKX3.1(R52C) (Fig. 1B). Western blotting confirmed that, as expected, neither NKX3.1(R52C) nor NKX3.1(S48A) induced IGFBP-3 protein expression in PC-3 cells (Fig. 1C).
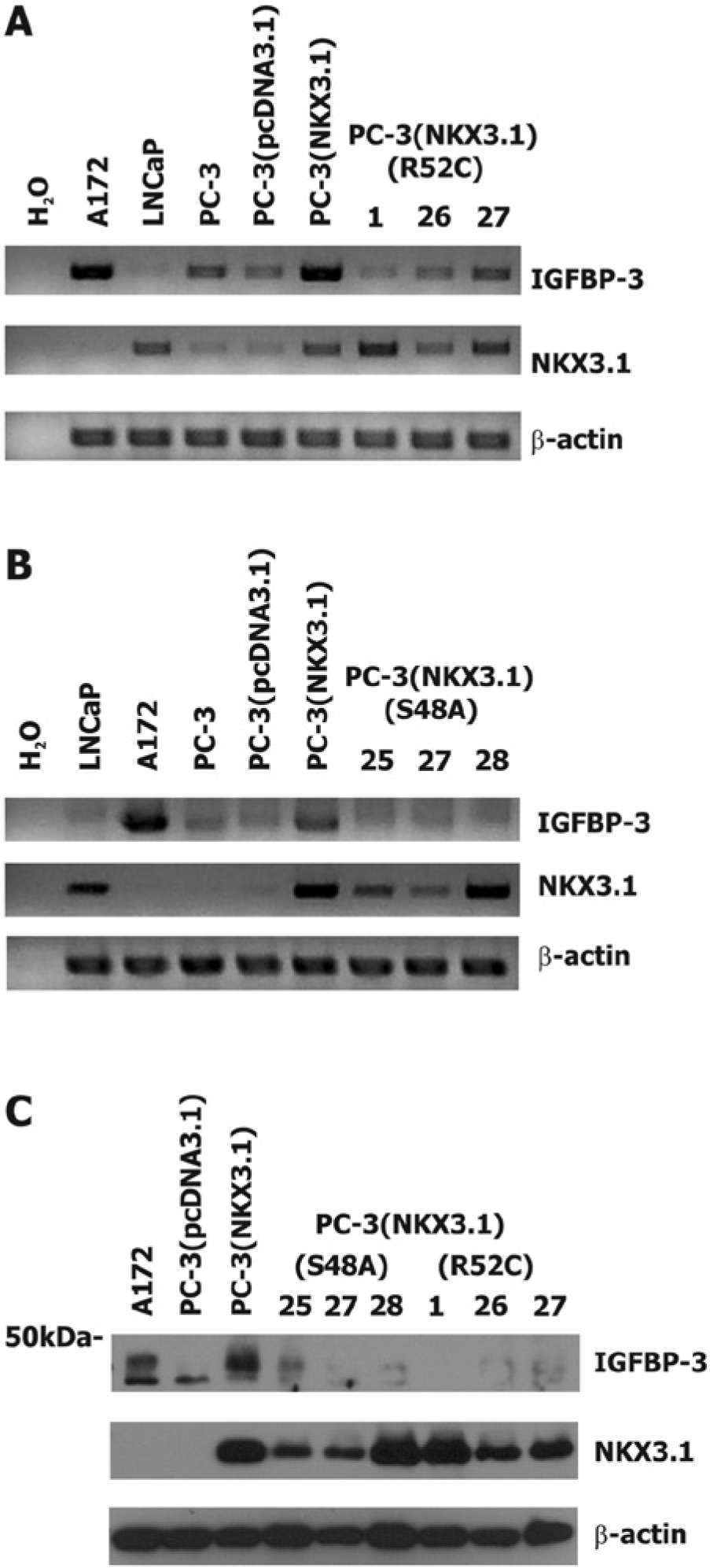
The effect of NKX3.1 variant proteins on IGFBP-3 expression in PC-3 cell clones. (
NKX3.1 expression attenuated IGFR-I activation in PC-3 cells via induced expression of IGFBP-3. The effect of NKX3.1 on IGFR-I activation was not seen when Long R-IGF-I, an IGFR-I ligand that does not bind to IGFBP-3 was used, or when cells were pretreated with IGFBP-3 siRNA. 21 In contrast, neither NKX3.1(R52C) nor NKX3.1(S48A) had an effect on IGFR-I signaling (Fig. 2A and B). Moreover, signaling downstream from IGFR-I to IRS-1 is attenuated by NKX3.1 expression, but not by either NKX3.1(R52C) or NKX3.1(S48A) (Fig. 2C).
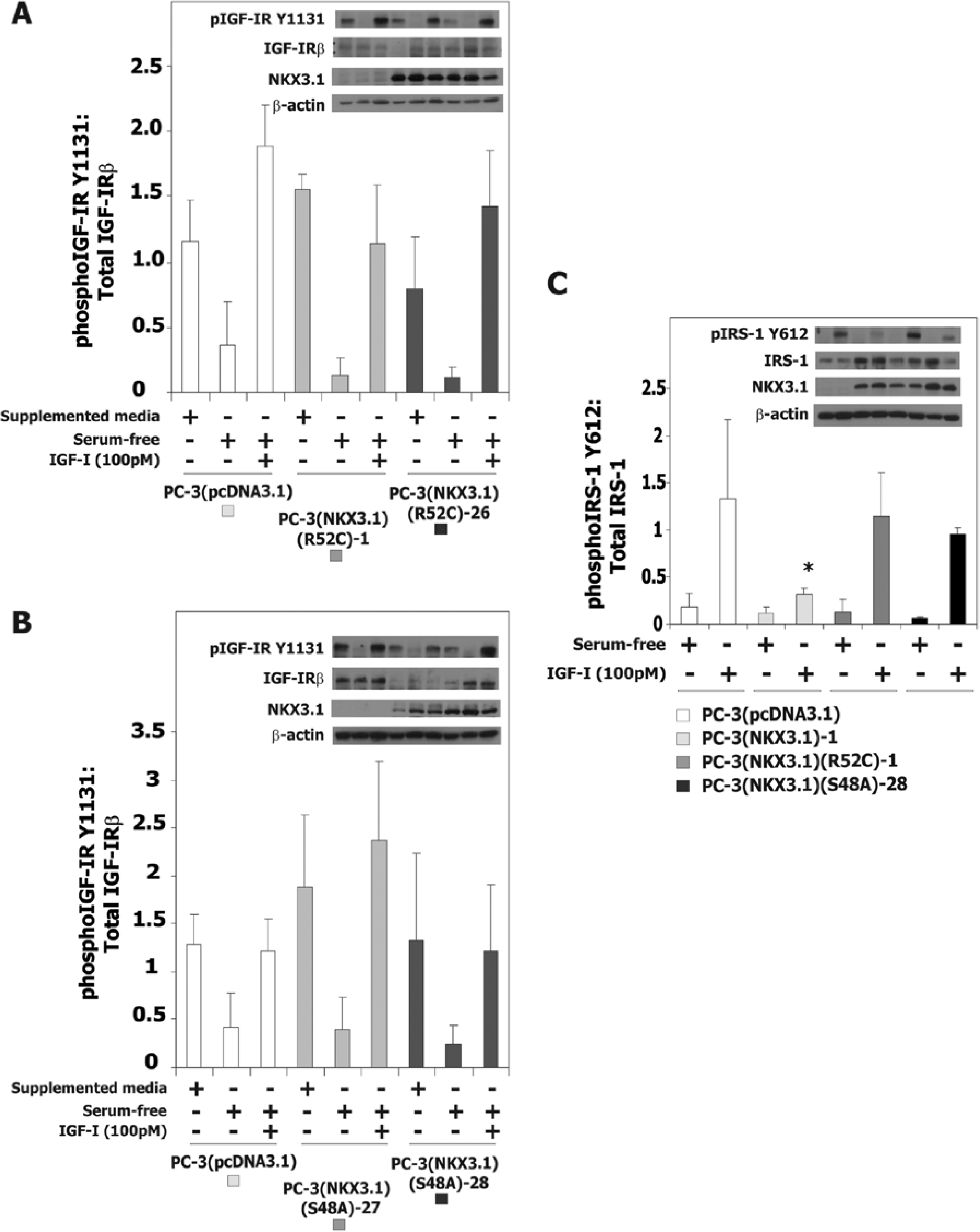
The effect of NKX3.1 variant proteins on IGF-IR activation in PC-3 cells. (
NKX3.1 suppresses cell proliferation in culture, an effect that is abrogated by siRNA to IGFBP-3. 21 PC-3 cells have a doubling time of approximately 24 hours that is extended to approximately 30 hours by expression of NKX3.1. Neither NKX3.1(R52C) nor NKX3.1(S48A) affected PC-3 cell doubling time (Table 1) despite levels of NKX3.1 protein expression comparable to levels of wild type protein that suppressed proliferation (Fig. 3).
Effect of NKX3.1 Expression on Cell Proliferation.
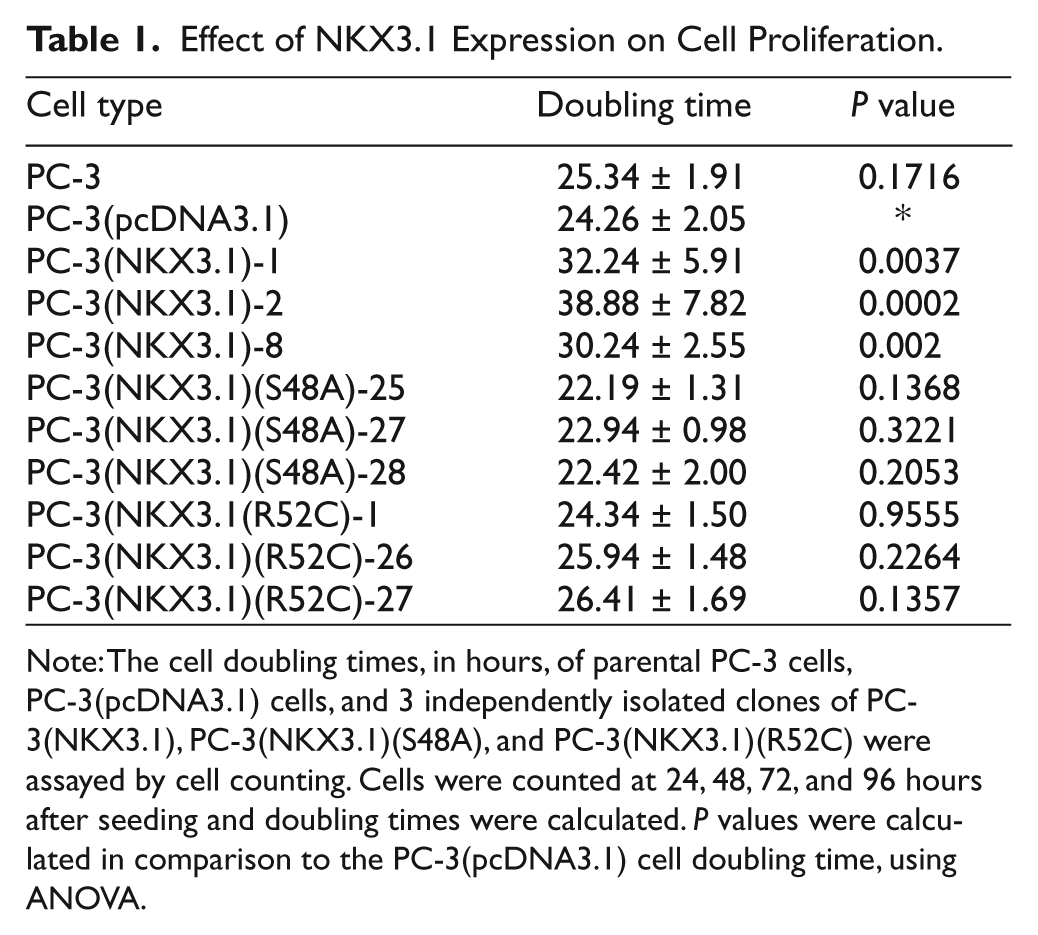
Note: The cell doubling times, in hours, of parental PC-3 cells, PC-3(pcDNA3.1) cells, and 3 independently isolated clones of PC-3(NKX3.1), PC-3(NKX3.1)(S48A), and PC-3(NKX3.1)(R52C) were assayed by cell counting. Cells were counted at 24, 48, 72, and 96 hours after seeding and doubling times were calculated. P values were calculated in comparison to the PC-3(pcDNA3.1) cell doubling time, using ANOVA.
Expression of NKX3.1 in derivative PC-3 cell clones. Panels from western blots of β-actin and NKX3.1 are shown from exponentially growing cultured cells from which equal amounts of total protein were loaded onto a gel. Numbers indicate identities of individual independently derived clones. A cell extract from PC-3 cells transfected with the empty vector is shown at the far left as a negative control.
NKX3.1(C154T) as a risk factor
Previously we had shown that among 1,253 cases and controls in the Physicians’ Health Study (PHS) NKX3.1(C154T) (rs2228013) that codes for NKX3.1(R52C) was a mild risk factor for prostate cancer. 20 We expanded this sample set to include cases from the PHS diagnosed more recently so that we now analyzed NKX3.1 genotypes from 937 prostate cancer cases and 1,086 age-matched controls. In this expanded sample set, the NKX3.1(C154T) allele was related to prostate cancer risk to a similar degree as we had previously published 20 (Table 2). Importantly, among men with prostate cancer diagnosed before age 70, the median age at diagnosis in the PHS, rs2228013 was significantly associated with higher risk of overall prostate cancer (relative risk [RR] = 1.55; 95% confidence interval [CI] = 1.04-2.31), Gleason <7 (RR = 1.71; 95% CI = 1.10-2.65), stage T1/T2 (RR = 1.67; 95% CI = 1.09-2.57), and a higher risk of lethal cancer (metastatic cancer at diagnosis or fatal cancer during follow-up; RR = 2.15; 95% CI = 1.00-4.63) (Table 3).
Odds Ratios of Prostate Cancer by NKX3.1 Genotype in the PHS.
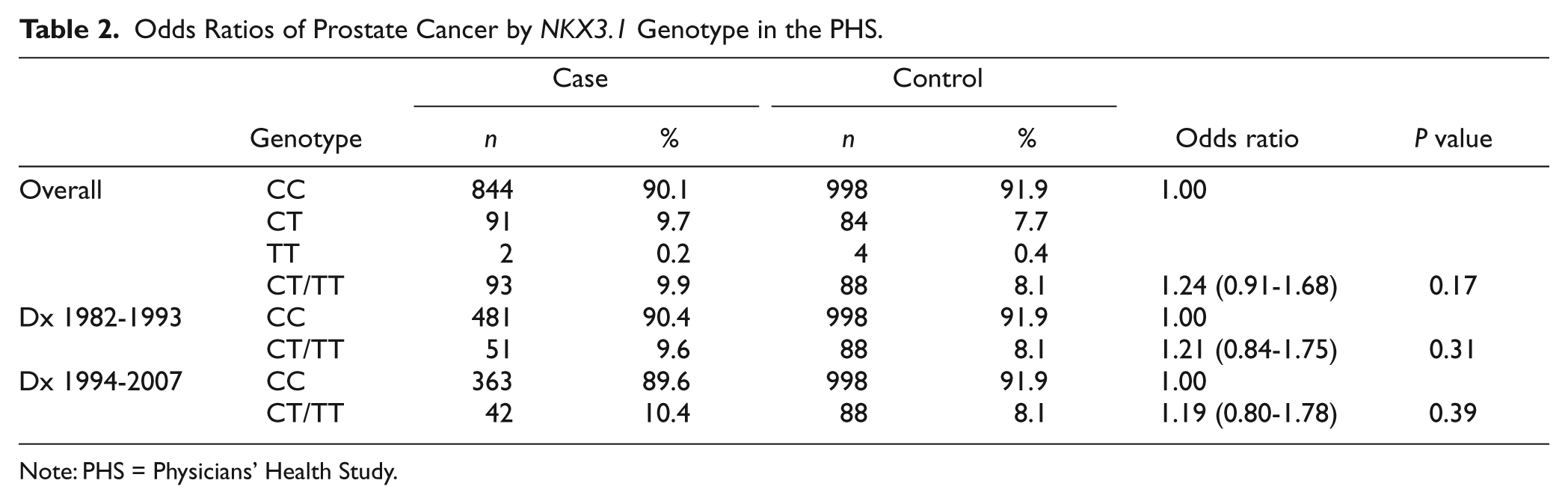
Note: PHS = Physicians’ Health Study.
Odds Ratios of Prostate Cancer by NKX3.1 Genotype Among Men Age <70 at Diagnosis: Results From the PHS.
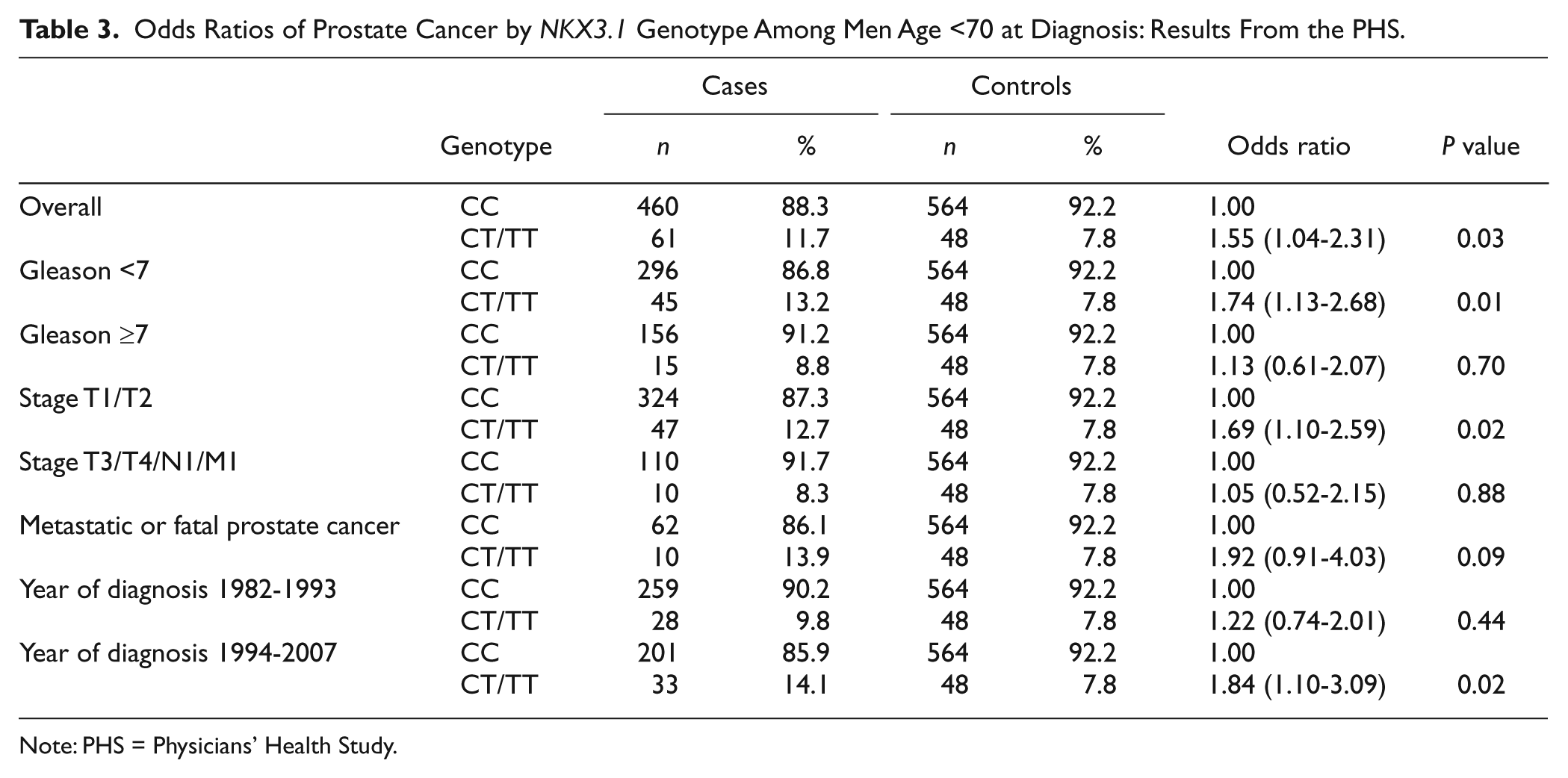
Note: PHS = Physicians’ Health Study.
Analysis of NKX3.1 genotype and serum IGF-I
Since NKX3.1 regulates local IGFBP-3 expression in prostate and IGFBP-3 attenuates IGF-I signaling, we asked whether NKX3.1 genotype influenced the effect of IGF-I on prostate cancer risk. From participants in the PHS, we analyzed 673 prostate cancer cases and 527 matched controls for which we were able to determine both NKX3.1 genotype and serum levels of IGF-I (Table 4). The majority of cases (69%) in this cohort had been ascertained prior to 1994 and thus had been diagnosed prior to the widespread use of serum prostate-specific antiven (PSA) for prostate cancer screening. Moreover, the blood samples were collected in 1982, prior to the diagnosis of prostate cancer in the cases. The frequency of rs2228013 among the control participants with IGF-I data was 6.83%, somewhat lower than the overall PHS controls (8.10%) and lower than the 11% we had originally found in our previous study. 20 Table 4 shows the frequency of cases and controls displayed by NKX3.1 genotype and distributed across 3 tertiles of plasma IGF-I levels. Among participants homozygous for wild type NKX3.1 there was no effect of plasma IGF-I on prostate cancer occurrence. However, among participants with NKX3.1 variant T allele we found a 2.5-fold higher risk of prostate cancer comparing the highest to the lowest tertile of plasma IGF-I levels. The positive trend was apparent only among prostate cancer cases diagnosed before 1993, but not among cases diagnosed 1994 and after. NKX3.1 genotype and serum IGF-I levels did not have a statistically significant interaction even among this subset probably due to the small sample size among T allele carriers (P for interaction = 0.2673).
Odds Ratios of Prostate Cancer by NKX3.1 Genotype and IGF-I Level in the Physicians’ Health Study.
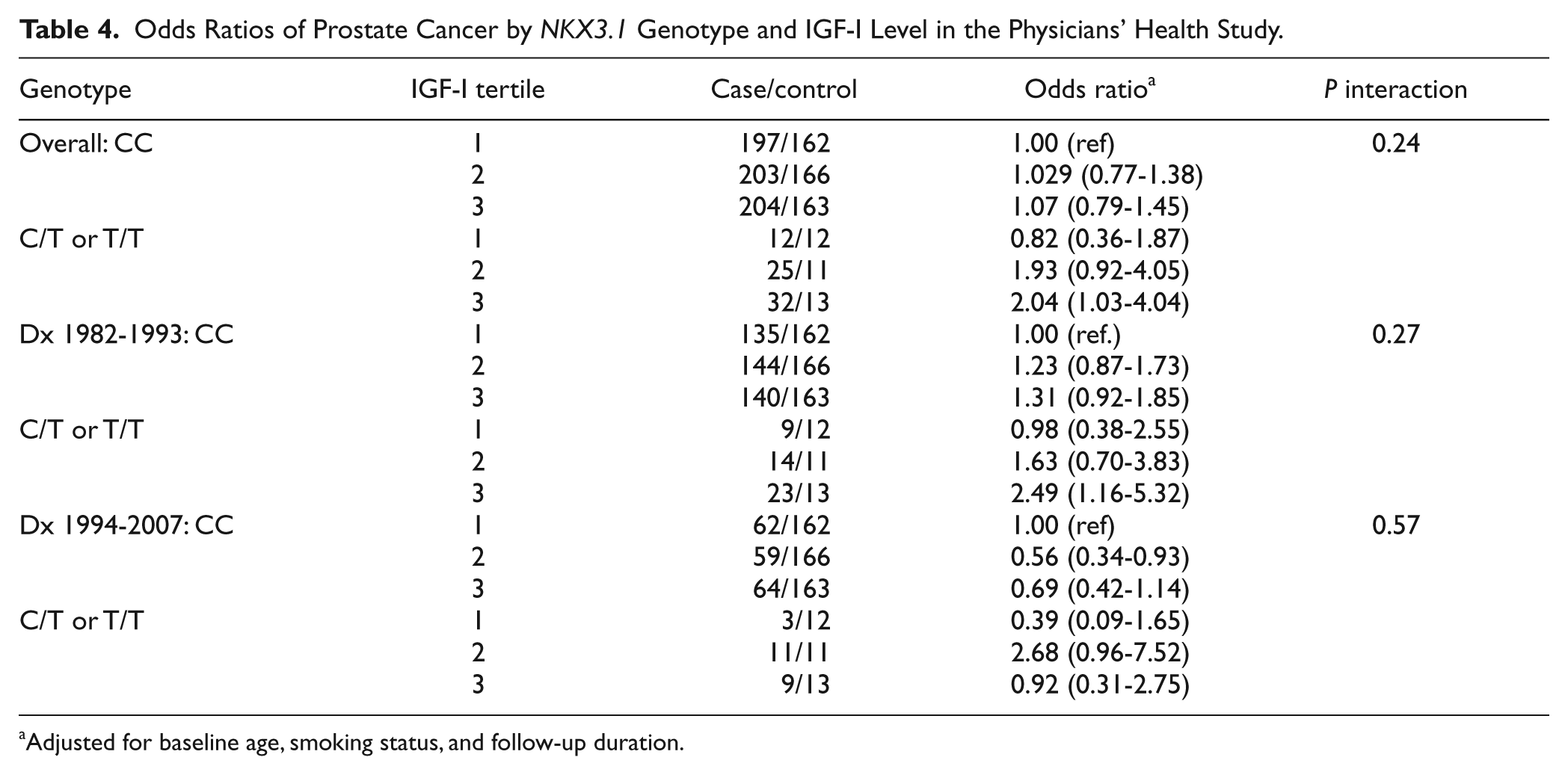
Adjusted for baseline age, smoking status, and follow-up duration.
We analyzed NKX3.1 genotype and serum IGF-I in a second group of cases and controls from the Risk Factors for Prostate Cancer Study of the Victoria Cancer Council (RFPCS). These cases were diagnosed between 1994 and 1997 and are therefore considered largely screen-detected. The characteristics of this population are shown in the right column of Table 5. Noteworthy is that no prostate cancer cases with Gleason score <5 were included in this cohort. In this population, rs2228013 was present in 10.4% of the participants. These screened cases had a trend toward a younger age of diagnosis, lower stage distribution, and lower grade distribution than the PHS cohort. Moreover, blood samples were drawn after the diagnosis of prostate cancer, not years prior as had been done with the PHS study samples. Among these participants there was no effect of NKX3.1 genotype on prostate cancer risk. There was also no interaction with levels of serum IGF-I (Table 6). However, the effect of serum IGF-I levels alone on prostate cancer risk was seen in this population (Table 7).
Characteristics of the Two Study Populations.
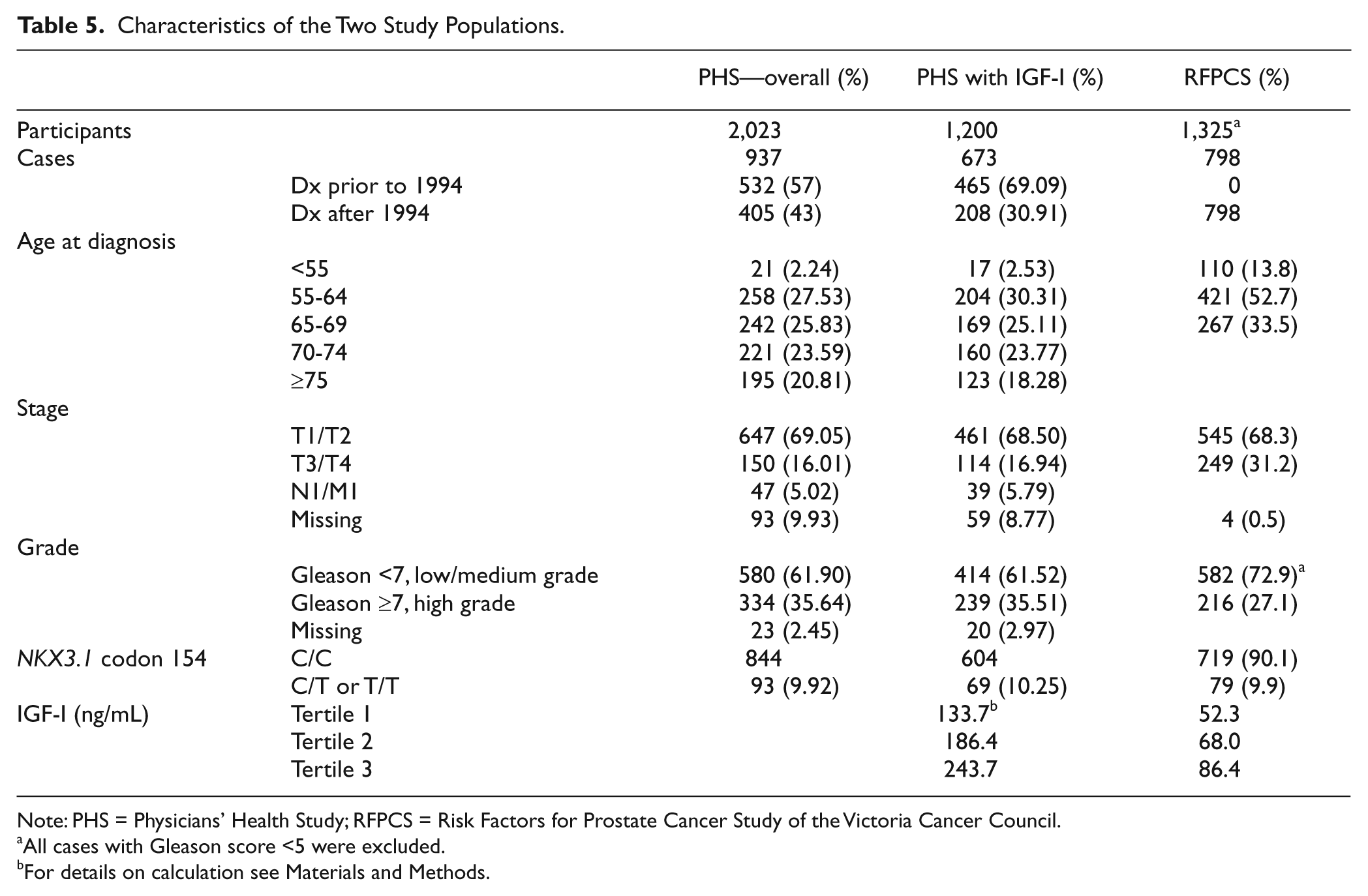
Note: PHS = Physicians’ Health Study; RFPCS = Risk Factors for Prostate Cancer Study of the Victoria Cancer Council.
All cases with Gleason score <5 were excluded.
For details on calculation see Materials and Methods.
NKX3.1 Genotype and Serum IGF-I Levels in Risk Factors for Prostate Cancer Study Participants.
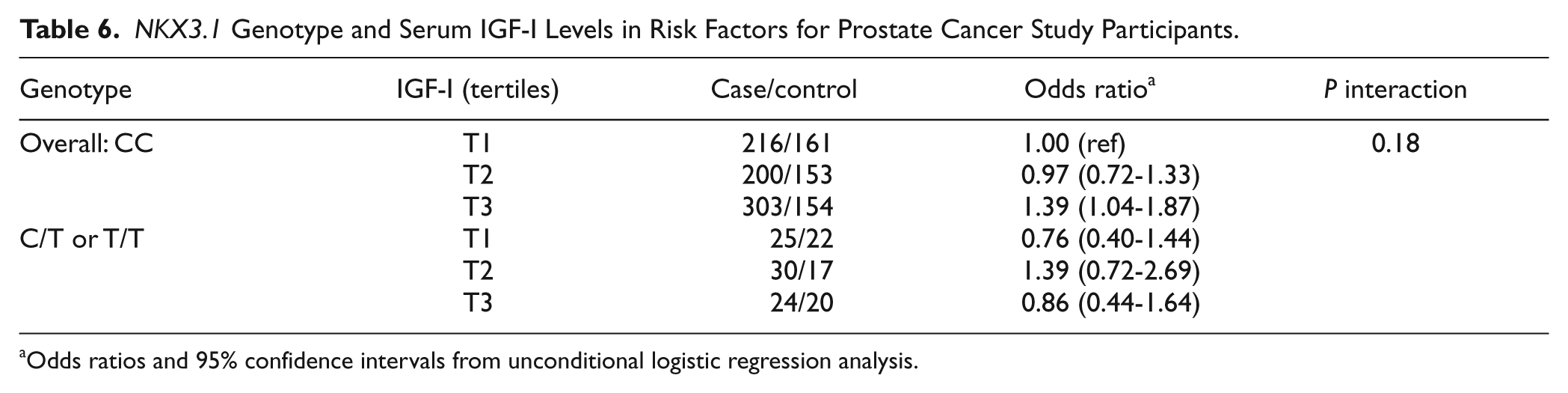
Odds ratios and 95% confidence intervals from unconditional logistic regression analysis.
Serum IGF-I Levels and Prostate Cancer Risk in Risk Factors for Prostate Cancer Study Participants.
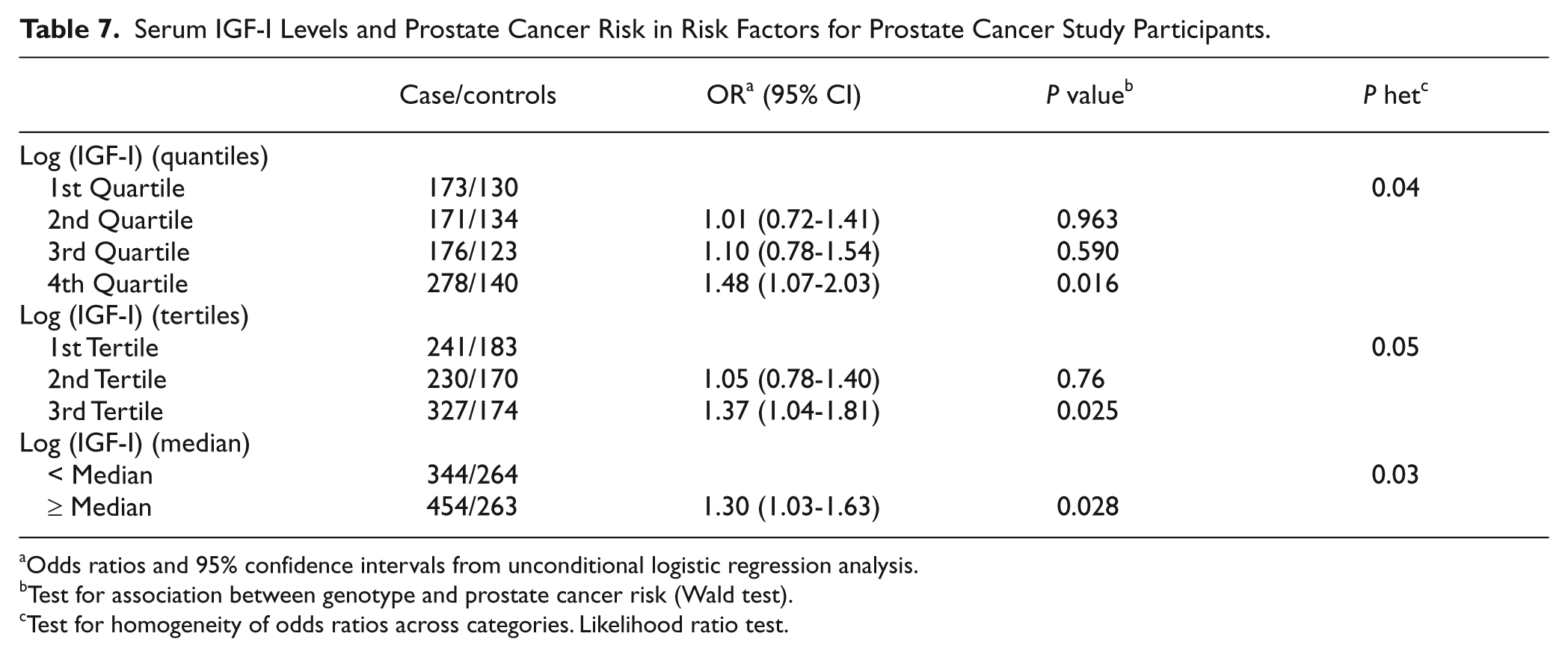
Odds ratios and 95% confidence intervals from unconditional logistic regression analysis.
Test for association between genotype and prostate cancer risk (Wald test).
Test for homogeneity of odds ratios across categories. Likelihood ratio test.
Discussion
NKX3.1 is important for prostate epithelial cell development, growth control, and differentiation.4,22 Murine Nkx3.1 is haploinsufficient and loss of a single allele manifests a phenotype similar to homozygous deletion, but with longer latency. 4 In early human prostate cancer we have found that NKX3.1 expression is down-regulated over a broad range of expression levels with a median expression in primary prostate cancer of 0.67, the level in adjacent normal cells. 3 In the course of determining the pathways of tumorigenesis that are affected by NKX3.1, we found that expression of IGFBP-3 in the prostate is downstream of NKX3.1 and that the effects of NKX3.1 on cell proliferation are mediated by IGFBP-3. In cultured cells NKX3.1 attenuates IGF-I signaling by activating IGFBP-3 expression. 21 Importantly, IGFBP-3 mRNA is among the genes most commonly down-regulated in prostate cancer tissues compared to normal prostate tissues, suggesting that its down-regulation may play a role in prostate cancer pathogenesis. 23
NKX3.1 exerts a broad range of effects on prostate epithelial cells. When NKX3.1 is down-regulated in the course of prostate carcinogenesis, 3 those effects vary with gene copy number as methylation due to NKX3.1 haploinsufficiency, well demonstrated by gene targeting experiments in mice.4,24 NKX3.1 is known to have a broad effect on transcriptional targets, both increasing and decreasing expression of many genes.21,25 However, our experiments with reporter constructs containing the cognate NKX3.1 DNA binding sequence have shown that promoter binding by NKX3.1 fails to activate transcription directly and only was observed to down-regulate gene expression. 26 NKX3.1 is able to cooperate with other transcription factors such as serum response factor to function as a coactivator and enhance transcription.27,28 The effect of NKX3.1 on gene transcription is therefore not well understood and may be dependent on a variety of factors including cell context and differentiation.
Previously we showed that both in cultured cells and in vivo there was a correlation between expression of NKX3.1 and IGFBP-3. In cultured cells NKX3.1 expression directly affects activation of the IGF-I receptor via control of IGFBP-3 expression. Moreover, proliferative effects of NKX3.1 in cultured cells are mediated via IGFBP-3 and its effect on IGF-I signaling. 21 In contrast, the interaction between NKX3.1 and IGF-I signaling in the prostate gland is likely to be more complex. As would have been predicted, conditional loss of Igf-I receptor in the prostate gland resulted in decreased proliferation and tumor suppression. 29 Moreover, prostate-specific transgenic expression of an Igf-I construct with attenuated Igfbp-3 binding resulted in prostatic hyperplasia but failed to cooperate with other carcinogenic signals to enhance transformation of prostate epithelial cells. 30 Thus, 2 independent experiments in mice suggested that Igf-I signaling could play a role in early stages of prostate carcinogenesis. In contrast, study of IGFBP family members in vivo is complicated by the observation that these proteins can compensate for one another. Thus, gene targeting of Igfbp-3 in the mouse is unlikely to define specifically the role of IGFBP-3 in prostate carcinogenesis.31,32 To demonstrate a phenotype from gene targeting, loss of Igfbp-3 was combined with loss of other members of the gene family. 31 Whether the same compensatory activation of IGF binding proteins occurs in human prostate epithelial cells that have reduced NKX3.1 expression was not determined. Last, detailed analysis of the effect of IGFBP-3 expression in human is complicated by the fact that IGFBP-3 is cleaved to multiple peptides with different degrees of activity and the rate and nature of the cleavage affects activity downstream of gene expression. 33
In the data we present here, the presence of a variant NKX3.1 allele was associated with prostate cancer risk in one prospective cohort but not in a second group of men assembled after the diagnosis of prostate cancer. Despite our observations from in vitro models that NKX3.1 blocks IGF-IR activation via IGFBP-3 we could not demonstrate a consistent interaction between NKX3.1 genotype and circulating levels of IGF-I in the 2 study populations. NKX3.1 genotype did affect Gleason score <7 tumors especially in men younger than 70. This observation is consistent with the notion that high-grade tumors are autonomous in their growth and less influenced by host factors, such as circulating IGF-I, or NKX3.1 expression. In contrast, tumors with lower Gleason scores may have greater dependence on such factors. It is also worth noting the NKX3.1 polymorphism was associated with an increased risk of lethal prostate cancer among those diagnosed prior to age 70.
Differences in the association between NKX3.1 genotype and prostate cancer risk in the 2 studies we analyzed may be due to the effect of overdiagnosis of prostate cancer. The PHS population includes a large fraction of cases diagnosed prior to mid-1990 and therefore prior to the adoption of widespread PSA screening for prostate cancer. On the other hand, the RFPCS includes only cases diagnosed between 1994 and 1997. Histologic foci of prostate cancer are concomitants of aging and are found in all men. PSA screening is a highly sensitive test that has caused an increase in the total number of cases diagnosed each year contributing to a substantial overdiagnosis. Some estimates identify as much as 42% or more of all cases as overdiagnosed. 34 Thus, many cases diagnosed during the PSA era may be biologically different from the bulk of those diagnosed before the adoption of screening on a large scale, when most diagnosed cases were clinically meaningful and carried a more serious prognosis. Ours is not the only study to find disparate results between case-control groups from the pre- and post-PSA screening era, and it is likely that PSA screening has altered the biology that is being analyzed in large case studies collected after the mid-1990s.35,36
The PHS and RFPCS also differed in the timing of blood collection that was used for serum IGF-I determinations. The PHS group was a nested case-control study, with prospectively collected samples. IGF-I levels were determined on samples collected before the diagnosis of cancer. In contrast, for the RFPCS case-control study, the blood was collected after diagnosis and perhaps after treatment had been initiated. Thus, in the case-control setting, one is always concerned that the disease, or treatment, or both could affect the blood levels.
Almost all prostate cancer initiation is accompanied by decreased expression of NKX3.1 protein. We do not know whether there is a threshold of NKX3.1 expression that predisposes cells to malignant transformation or whether the risk of malignant transformation is related to the level of NKX3.1. Consistent with the role of NKX3.1 as a tumor suppressor, a T164A missense mutation in the NKX3.1 homeodomain that reduced DNA binding by 95% was found to cosegregate with early prostate cancer in a family. 9 Phenotypic effects resulting from reduced levels of NKX3.1 protein may reflect the fact that other members of the NK protein family are also haploinsufficient. For example, both missense and truncation mutations that cause loss of NKX2.5 protein are autosomal dominant determinants of congenital cardiac abnormalities.37,38 Our data and work with gene targeted mice suggest that tumor suppression by NKX3.1 correlates with protein expression levels and that optimal function of the protein requires native expression from 2 intact NKX3.1 alleles. Analysis of downstream targets of Nkx3.1 identified a number of genes related to differentiation but did not identify Igfbp-3. 39 We, on the other hand, showed that prostatic Igfbp-3 mRNA levels correlate with Nkx3.1 gene copy number in gene-targeted mice. 21 Further studies will identify additional NKX3.1 targets and elucidate their role in prostate cancer suppression and, perhaps, prevention.
Materials and Methods
Cell culture and reagents
The prostate cancer cell lines PC-3 and LNCaP and the A172 human glioblastoma cell line were obtained from the American Type Culture Collection (Rockville, MD). PC-3 and A172 cell lines were grown as previously described. 21
Plasmids and transfection full length NKX3.1, NKX3.1 (R52C), and NKX3.1(S48A) were expressed from constructs in the mammalian expression vector, pcDNA3.1 (Invitrogen, Carlsbad, CA) as previously described. 20 Transient and stable transfections were carried out as described earlier. 21
Western blot analysis
Immunoblotting was done as previously described. 21 Antibody reagents were as follows: β-actin (Sigma, St. Louis, MO) 1:10,000; NKX3.1 21:2,000; IGFBP-3 (sc-9028, Santa Cruz Biotechnology, Santa Cruz, CA) 1:8,000 at 4°C overnight, followed by 3 washes in phosphate-buffered saline. Horseradish peroxidase conjugated goat-anti-rabbit and goat-anti-mouse (ImmunoPure antibodies, Pierce Biotechnology, Rockford, IL) secondary antibodies in 1% milk or 1% bovine serum albumin were applied for 1 hour at room temperature. Signal detection was performed with Super-Signal West Pico Chemiluminescent Substrate (Pierce Biotechnology).
Reverse transcriptase PCR analysis
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Venlo, the Netherlands) and cells were homogenized using the Qiashredder (Qiagen) method. A total of 125 to 250 ng of RNA was added to the RT-PCR master mix from One-step RT-PCR kit (Qiagen) (includes 5× buffer, DNTPs, and Taq polymerase). The following primers were used in the RT-PCR reactions: β-Actin (Fwd 5′-GGC CAC GGC TGC TTC-3′ and Rev 5′-GTT GGC GTA CAG GTC TTT GC-3′); NKX3.1 (Fwd 5′-GCC GCA CGA GCA GCC AGA GAC A-3′ and Rev 5′-TTC AGG GCC GGC AAA GAG GAG TG-3′); IGFBP-3 (Fwd 5′-CGC CAG CTC CAG GAA ATG-3′ and Rev 5′-GCA TGC CCT TTC TTG ATG ATG-3′); IGFBP-4 (Fwd 5′-TTA GCC CAA GAG GTC TGA GC-3′ and Rev 5′-CTG TGC TTC AAG TCT TCC TTT G-3′); Lamin A/C (Fwd 5′-AAC TTC AGG ATG AGA TGC TGC G-3′ and Rev 5′-GTC CAG AAG CTC CTG GTA CTC GT-3′). RT-PCR was performed in a Techne Techgene PCR machine; 30 minutes at 50°; 15 minutes at 94°; 22 to 30 cycles of 30 seconds to 1 minute at 94°, 30 seconds to 1 minute at melting temperatures of 55° to 65°, and 30 seconds to 1 minute at 72°; followed by 15 minutes at 72°. Samples were stored on ice and mixed with 10× Blue Juice gel loading buffer (Invitrogen) and run on a 1.5% agarose gel containing 0.1 µg/mL ethidium bromide in TAE buffer. Gels were imaged on a luminometer and recorded using a Kodak 1D digital camera.
IGF-IR activation and signaling
IGF-IR signaling was assayed as described previously. 20 Western blot analysis was completed as described above using anti-IGF-IR (#3027, Cell Signaling, Beverly, MA), anti-phospho-IGF-I Receptor (Tyr1131) (#3021, Cell Signaling), anti-IRS-1 (06-248, Upstate, Placid Lake, NY), anti-phospho-IRS-1(Y612) (44-816G, Biosource, Carlsbad, CA) primary antibodies. Bands were quantified by Scion Imager software and P-values were assessed from triplicate experiments by t-test analysis using Prism Graphpad software (* indicates P-value < 0.05).
Cell proliferation assay
PC-3 and derivative cell lines were seeded in triplicate in 96-well plates at a concentration of 4,000 cells per well in IMEM containing 10% fetal bovine serum (PC-3) or 10% fetal bovine serum plus 1.2 mg/mL G418 (PC-3(pcDNA3.1) and PC-3(NKX3.1)) and incubated for 24 hours at 37°C. At 24, 48, 72, and 96 hours after seeding, wells were trypsinized, suspended in IMEM, and immediately counted in a Beckman Coulter Z1 cell counter. Doubling times were calculated using Microsoft Excel, and P-values were calculated by ANOVA.
Study cohorts
Physicians’ Health Study: The participants of the Physicians’ Health Study (PHS) who comprised the prostate cancer cases and controls have been previously described in articles describing the effect of serum IGF-I 22 and NKX3.1 genotype 23 on prostate cancer risk. The current analysis includes 937 cases and 1,086 controls, using a prospective nested case-control design. Of the cases, 580 had Gleason score <7, 334 had Gleason score ≥7, and 23 cases had unknown Gleason scores. Among the cases, 647 were T1/T2 and 197 were locally advanced or metastatic at presentation, with the remaining 93 cases of unknown stage. Median age of cases at diagnosis was 70 years.
Risk factors for prostate cancer study
The Risk Factors for Prostate Cancer Study (RFPCS) is a population-based case-control study of prostate cancer in Australia. Prior approval of the study protocol was obtained from all relevant hospital and cancer registry human research ethics committees in Melbourne and Perth. Eligible cases comprised male residents of Melbourne and Perth diagnosed between 1994 and 1997 and recorded in the population-based cancer registries with a histopathology confirmed diagnosis of adenocarcinoma of the prostate (International Classification of Diseases, 9th revision, rubric 185), excluding well-differentiated tumors (defined as low grade, i.e., those with Gleason scores <5). Cases had to be <70 years of age at diagnosis. Controls were randomly selected from men on the current state electoral rolls and were frequency matched to the predicted age distribution of the cases in a ratio of one control per case. Potential controls were matched against the cancer registries at the time of recruitment to exclude men with a known history of prostate cancer. Controls were identified and interviewed contemporaneously with the cases over the period 1994 to 1997. The cohort comprised >95% of Caucasians. The lack of racial diversity is not critical to this analysis since the frequency of the NKX3.1 polymorphism does not vary between races. 23 A total of 798 cases and 527 controls were analyzed. Of the total cases, 582 were described as low-to-moderate grade and 216 as high grade (Gleason score >7 or poorly differentiated or undifferentiated tumors). Among the cases, 545 were T1/T2 and 249 were locally advanced or metastatic at presentation, with the remaining 4 cases of unknown stage. Median age for cases and controls was 62 years.
Serum IGF-I assay
Serum IGF-I levels were determined by ELISA with reagents from Beckman Coulter (DSL, Webster, TX). All assays were performed in the laboratory of one of the authors (MJP). Reliability of laboratory assays of plasma levels are always checked before measuring the real samples. The mean intrapair coefficients of variation for blinded duplicate quality control samples were 2.6% for IGF-I, and the long-term intraperson correlation coefficient for these biomarkers was 0.66 for IGF-I (3 years apart). 14 Cases and controls were separated by tertile of serum IGF-I levels. The median values for the IGF-I levels in the tertiles from the controls in each study are reported. For the PHS, the IGF-I measurements were made at 4 different times when batches of 92, 257, 73, and 105 control samples were assayed. Batch-specific cutoff points were used for tertiles, and overall median values are reported as the mean of the 3 median values. Inclusion of the IGF-I values for the cases as well as controls changed the overall means by less than 1% for each tertile.
Statistical analysis
For the PHS, the baseline characteristics and information at prostate cancer diagnosis for all the 937 cases and 1,086 controls and for the subgroup of 673 cases and 527 controls with both the genotype data and plasma IGF-I levels are presented in Table 5. Using Pearson’s goodness-of-fit test, the NKX3.1 SNP does not violated Hardy-Weinberg equilibrium (P > 0.05). Because of the low prevalence of the variant T allele, SNP was analyzed under a dominant model for both main effect analysis and the test of interactions. Cases and controls were matched by age and follow-up duration, but not by race. Because excluding non-Caucasians or conducting subgroup analysis led to losing some case-control pairs, we used an unconditional logistic regression model, to assess the risk of incident prostate cancer according to genotype, adjusting for the matching factors (age at randomization, smoking status, and follow-up time). We also conducted a subgroup analysis, comparing cases with Gleason score ≥8 or clinically advanced stage (T3, T4, N1, or M1) to all controls.
We first conducted case-control analysis assessing the overall association of the NKX3.1 polymorphism with risk of developing prostate cancer among all the 937 cases and 1,086 controls. We also evaluated the associations separately by Gleason grade (<7 vs. ≥7), by clinical stage (localized T1/T2 vs. advanced T3/T4/N1/M1), by fatal prostate cancer as outcome, and by year of the cancer diagnose (1982-1994 vs. 1997-2007) (Table 2). These analyses were then repeated by median age at diagnosis (<70 vs. ≥70 years). Finally, we assessed interaction between baseline plasma IGF-I levels and the NKX3.1 polymorphism (Table 4). We then assessed this interaction in the RFPC study using the same strategy. All statistics were calculated using SAS (version 9.1.3; SAS Institute Inc, Cary, NC), with a 2-sided significance level of 0.05.
Footnotes
Acknowledgements
We thank Dallas English and John Hopper, principal investigators of the Risk Factors for Prostate Cancer Study, for their contributions to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIEHS Grant ES09888 to EPG and by DOD Grants W81XWH-07-1-0263 to EPG and PC-05-0590 to EM. PHS was supported by NIH Grants CA42182, CA90598 and DOD Grants PC050569, CA097193. The RFPC study was supported by grants from the Australian National Health and Medical Research Council (#504700 and #504702) and by infrastructure from the Cancer Council Victoria.