
Abstract
Glioblastoma multiformes (GBMs) are extensively heterogeneous at both cellular and molecular levels. Current therapeutic strategies include targeting of key signaling molecules using pharmacological inhibitors in combination with genotoxic agents such as temozolomide. In spite of all efforts, the prognosis of glioma patients remains dismal. Therefore, a proper understanding of individual molecular pathways responsible for the progression of GBM is necessary. The epidermal growth factor receptor (EGFR) pathway is probably the most significant signaling pathway clinically implicated in glioma. Not surprisingly, anti-EGFR therapies mostly prevail for therapeutic purposes. The Wnt/β-catenin pathway is well implicated in multiple tumors; however, its role in glioma has only recently started to emerge. We give a concise account of the current understanding of the role of both these pathways in glioma. Last, taking evidences from a limited literature, we outline a number of points where these pathways intersect each other and put forward the possibility of combinatorially targeting them for treatment of glioma.
Introduction
Malignancies of the central nervous system are generally classified as meningiomas, oligodendrogliomas, and astrocytomas. Astrocytomas are further subdivided into pilocytic (grade I), diffuse (grade II), anaplastic (grade III), and glioblastoma multiforme (GBM; grade IV). GBM represents the most common and aggressive primary brain tumor with a median survival of approximately 12 months after diagnosis. Despite aggressive therapeutic modalities including surgical resections followed by radiotherapy and the most up-to-date chemotherapeutic regimen, the prognostic statistics for GBM patients is discouraging. Treatment of GBM demands a multipronged attack, based on a comprehensive understanding of the molecular principles governing the growth and survival of malignant cells. The biggest challenges in GBM treatment are (a) the extremely narrow window for operative approaches, which is mostly palliative and rarely curative; and (b) the blood–brain barrier, which poses a major challenge in successful drug delivery. Similarly, radiation therapy alone for GBM patients is unlikely to be curative. Therefore, most investigators and clinicians believe that development of better chemotherapeutic agents targeting multiple and specific aspects of glioma holds the best promise for patient outcome. 1
More than 2 decades of research has identified important molecular events, such as overactivation of the EGFR and PI3K (phosphatidylinositide 3 kinase)/Akt pathways and inactivation of the p53/Rb pathways to be important contributors for glioma development. These alterations facilitate cell survival, proliferation, and migration and thus are crucial for tumorigenesis. 2 Another important factor that has recently emerged to be a significant determinant of glioma initiation and progression is the canonical Wnt/β-catenin signaling pathway. Apart from a direct survival advantage provided by Wnt signaling to the tumor cells, 3 its role in stem cell biology is crucial. 4 The widespread attention the concept of cancer stem cells (CSCs) has gained in the last few years makes Wnt signaling an attractive target for glioma therapy. 5
However, it is surprising that very little work has been devoted to studying the crosstalk between these 2 signaling pathways in the context of glioma. Although published evidences are limited, in the last part of the review we focus on the collusion of EGFR and canonical Wnt pathways in glioma and try to point toward the possibility of combinatorially targeting them for the treatment of glioma.
EGFR and Wnt Are Important Survival and Growth Signaling Pathways
EGFR Signaling
The epidermal growth factor receptor (EGFR; also ErbB1 or Her1) belongs to the ErbB family consisting of 4 receptors (ErbB or Her 1-4) and 13 polypeptide extracellular ligands with a conserved EGF domain. Structurally, this 170 kDa protein consists of an extracellular ligand-binding domain, a hydrophobic domain spanning the membrane, and a cytosolic tyrosine kinase domain. On binding of ligand to the extracellular domain, the receptor dimerizes (homo or heterodimers), leading to phosphorylation of the cytosolic tail (autophosphorylation and/or cross-phosphorylation) with subsequent activation of several downstream kinases and finally transcription factors. 6 On activation of the EGFR, various adaptor and effector molecules like Shc (Src homology 2 containing transforming protein); Grb2 and Grb7 (growth factor receptor-bound protein); c-Crk-p38; PLC-γ (phospholipase C-γ); kinases—JAKs (Janus kinases), Src (c-Src tyrosine kinase), and PI3K; protein tyrosine phosphatases—SHP1 and 2; E3 ligase Cbl; and transcription factors like STAT (signal transducer and activator of transcription) 1, 3, and 5 are activated, triggering several signaling cascades—Ras/Raf (rat sarcoma/rapidly accelerated fibrosarcoma), MAPK (mitogen activated protein kinase), PKC (protein kinase C), PI3K/PKB (protein kinase B), JAK/STAT, and so on—which results in the activation of Erk (extracellular signal-regulated kinase), JNK (c-Jun N-terminal kinase), p38-MAPK, NF-κB (nuclear factor κ-light-chain-enhancer of activated B-cells), transcription factors like c-Myc, and so on, that ultimately regulate the expression of genes involved in proliferation, survival, differentiation, and migration7-9 (see Fig. 1).
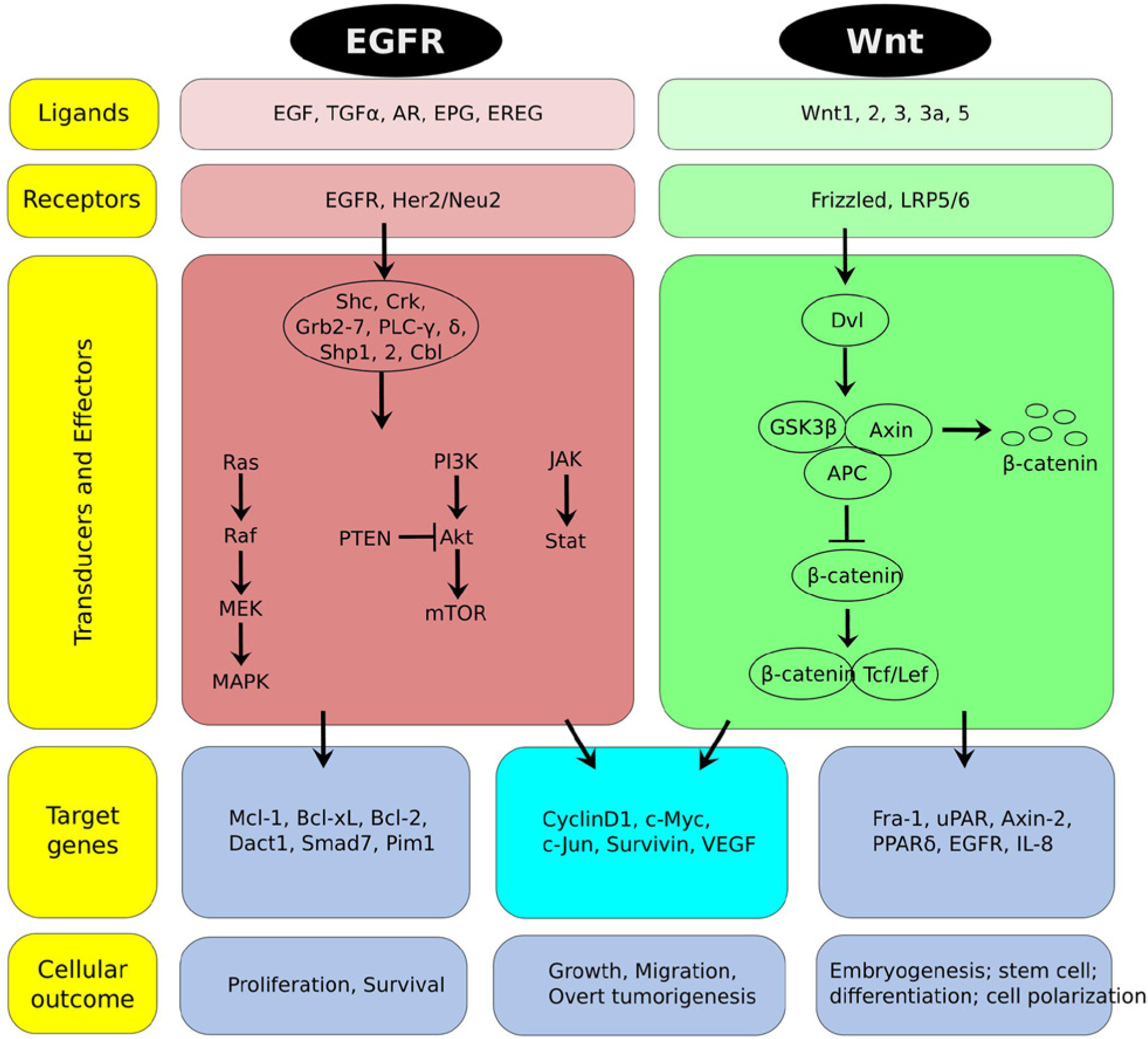
An overview of EGFR and Wnt/β-catenin signaling. EGFR signaling pathway. On binding of specific ligands, the EGF receptor dimerizes, activating an array of molecules including kinases, Grbs, and so on, initiating several downstream signaling cascades—MAPK, PI3K/Akt, JAK/STAT—which upregulate multiple transcription factors, anti-apoptotic and prosurvival proteins pushing the cells toward prolonged survival, proliferation, migration, and tumorigenesis. Wnt signaling pathway. In the absence of Wnt, β-catenin is captured by APC and Axin and forms a destruction complex in which its phosphorylation by the kinases CK1α and GSK3β is facilitated, which, in turn, allows binding of β-TrCP, which subsequently mediates the proteasomal degradation of β-catenin. On binding of the Wnt ligand to its cognate receptors Fzd and LRP5/6 triggers the formation of Dvl–Fzd complexes and the phosphorylation of LRP by CK1γ, facilitating relocation of Axin to the membrane and inactivation of the destruction complex. This allows β-catenin to accumulate and enter the nucleus, where it interacts with members of the Tcf/Lef family and exerts its transcriptional activity. For a more comprehensive and detailed account of these pathways, the readers should consult excellent reviews available in the literature.
EGFR phosphorylated at residues Y1068 and Y1086 can recruit Grb2 directly or indirectly via tyrosine phosphorylated Shc leading to translocation of Grb2/Sos complex to the membrane, where it activates Ras proteins, which in turn induce Raf kinases, MEKs and MAPKs, and Erk1/2 nuclear transport leading to cell proliferation by activating transcription factors like c-Myc and RSK (ribosomal S6 kinase). 10 Recently, EGFR-mediated MAPK signaling has been shown to attenuate the Groucho-mediated gene repression, establishing a node for crosstalk between the EGFR, Notch, Wnt, and TGF-β (transforming growth factor-β) signaling pathways.11-13 EGFR-mediated PI3K/PDK1(phosphoinositide-dependent protein kinase 1)/Akt pathway plays a crucial role in sustained cell survival and proliferation. Phospho-EGFR-Y920 can dock PI3K, converting PIP2 to PIP3 (phosphatidyl inositol (4,5) bisphosphate to (3,4,5) triphosphate), phosphorylating Akt, which inactivates the apoptotic cascade via Bad (Bcl2 associated death factor) and caspase-9. 14 In PTEN (phosphatase and tensin homolog) null or mutated GBMs also, Akt is constitutively activated, thus providing survival signals. 15 STATs are very important transcription factors downstream of EGFR responsible for cellular transformation and migration.16,17 JAK dependent activation of STAT1 and STAT3 or JAK-independent activation of STAT 5b (docking at EGFR Y845) 18 leads to their dimerization (homo and heterodimers) and nuclear translocation, where they act as transcription factors for several growth promoting genes like c-jun, c-fos, and so on.19,20 Interestingly, inhibiting the activity of PI3K and in turn Akt significantly increases the DNA-binding activity of STAT3 in U87MG and D54 cells. While considering this apparent contradiction, it should be noted that the major oncogenic role of STAT3 is because of the phosphorylation at Y705 while the negative regulation being discussed here concerns S727 and the other Ser/Thr residues at the C-terminal end of STAT3. Also, the phosphorylation at S727 has opposing roles, both oncogenic and tumor suppressive, in a context-specific manner. The finding, however, indicates a crosstalk between the 2 survival pathways, which decides the cell fate via a common player, Mcl-1 in this case. 21 Regulation of angiogenesis and metastasis are important functions of the EGFR. EGFR promotes angiogenesis by upregulating VEGF (vascular endothelial growth factor) and MMPs (matrix metalloproteinases). 22 Also, phosphorylated EGFR (Y992) directly interacts with and activates PLC-γ, which regulates actin cytoskeletal reorganization and hence cell motility. 23 Thus, in general, the multifaceted nature of the EGFR signaling in cancer can be clearly understood, which is being exploited as therapy, and some of the molecules in the EGFR pathway are important targets for drug development.
The EGFR gene is a major target for alterations in glioma
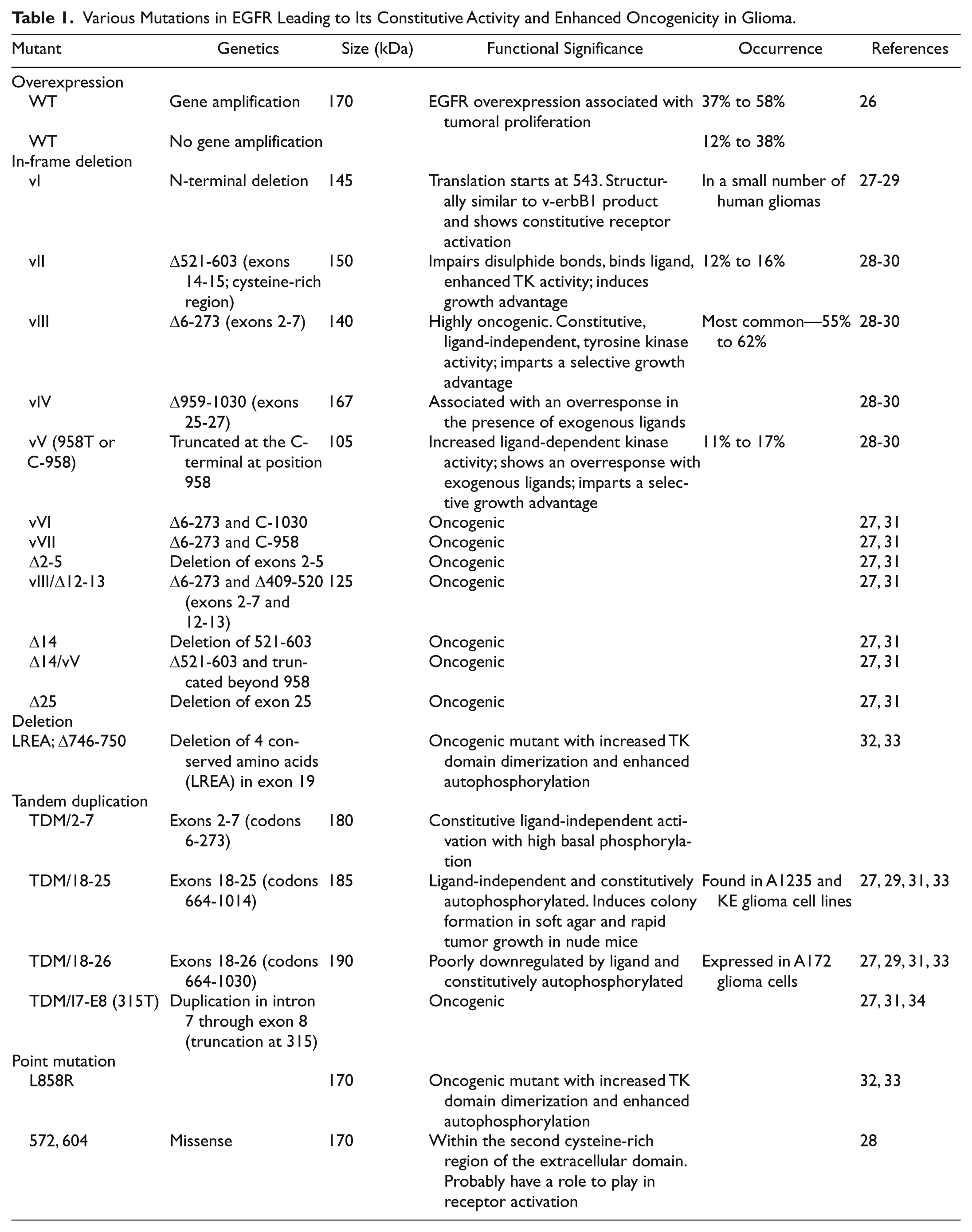
EGFR is one of the major genetic factors affecting the pathogenesis and prognosis of GBM. Genetic amplification, elevated expression, and mutation of EGFR have been widely implicated in various cancers and its role in influencing multiple aspects of tumorigenesis is well documented. Almost 50% to 60% of GBMs are genetically marked with overactivated EGFR and multiple functionally distinct forms of EGFR have been associated with glioma (see Table 1). The most common mutation is EGFRvIII (in-frame deletion spanning exons 2-7, imparting ligand-independent activation and high oncogenicity). In primary GBMs, EGFR aberrations are accompanied with mutations in tumor suppressors p16Ink4a, p19Arf, and PTEN, constitutively activating several oncogenes like STATs, Akt, Erk1/2, and so on, while secondary GBMs are mostly associated with mutation/deletions in p53, deregulating the cell cycle and also aberrant activation of PDGFR (platelet-derived growth factor receptor) pathway and IDH1 (isocitrate dehydrogenase 1) mutations. In PTEN null/mutated GBMs, Akt is constitutively activated, providing survival signals.24,25
Various Mutations in EGFR Leading to Its Constitutive Activity and Enhanced Oncogenicity in Glioma.
Alternate mechanisms for activating EGFR in glioma
Apart from EGFR itself, a number of different mechanisms have been used by cancer cells to overactivate EGFR signaling. For example, the ligands of receptor tyrosine kinase (RTK) family such as PDGF, TGF-α, and EGF are often overexpressed in glioma leading to positive feedback autocrine loops.35,36 Also, the need for EGFR overactivation can be bypassed or complemented by activating other RTK signaling such as PDGFR or by functionally obliterating important negative regulators such as PTEN. As a result, several crucial downstream players of EGFR pathway such as Akt and Ras gain constitutive activity.37-39 Similarly, in a subset of GBM, monoallelic loss of NF-κB has been shown to cooperate with EGFR to negatively affect patient prognosis. 40 Furthermore, observation that the bispecific EGFR/HER2 inhibitor AEE788 displays radiosensitizing effect on EGFR overexpressing cells suggests that at least in certain treatment modalities the responsiveness of therapy depends on the relative ratio between EGFR and HER2 and not on any individual receptor.
EGFR is involved in sustenance of glioma stem cells (GSCs)
EGFR signaling regulates proliferation and migration of neural stem cells (NSCs). The balance between EGFR signaling and a classical stem-cell signaling like Notch decides the proportion of cells in the stem cell niche. 41 Hence, the tumor microenvironment is a crucial factor in the maintenance of tumor initiating and self-renewing capacity of the CSCs. Like normal NSCs, the GSCs probably maintain their undifferentiated state and multipotency due to regulation of signaling in their niche. 42 Constitutively activated EGFR signaling (EGFRvIII) in GBM leads to activation of Akt, which can directly phosphorylate Smad5, which in turn leads to overexpression of ID3 (inducer of differentiation 3) and the cytokines like GRO1 (growth regulated alpha protein), interleukins (IL-6 and IL-8) supporting proliferation, angiogenesis, and acquisition of GSC-like characteristics. This signaling cascade imparts tumorosphere-forming ability to the GSCs and can be inhibited by EGFR inhibitors. 43
The growing tumor mass around the GSCs are often exposed to several stresses like hypoxia, low pH, nutrient deprivation at later stages, 44 and over 2 decades of research has established hypoxia to promote tumor cell proliferation, survival, angiogenesis, and motility, by triggering the adaptive transcriptional mechanisms. During hypoxic stress, the stem cell properties of neural progenitors (NPCs) are enhanced, 45 and also CD133+ (cluster of differentiation 133 positive) GSCs proliferate. 46 One of the key factors induced by hypoxia is HIF-1α (hypoxia-inducible factor 1α), 47 which is also regulated by EGF and other growth promoting signals like bFGF (basic fibroblast growth factor), PDGF, IGF (insulin-like growth factor), although to a lesser extent. Studies have revealed the crucial role of EGFR signaling cascade in the maintenance of GSCs, wherein EGF is capable of promoting sphere formation and self-renewal of the CD133+ subpopulation. This can be inhibited by specific pharmacological inhibitors like Gefitinib and AG1478 (tyrosine kinases), PD98059 (Erk1/2, MAPK), LY294002 (PI3K), and Rapamycin (mTOR). 48 Hypoxia inhibits differentiation of these CD133+ GSCs and supports their proliferation mainly by upregulating HIF-1α and VEGF (acting as a paracrine signal) and also by activating the EGFR downstream signaling molecules PI3K/Akt and Erk1/2, which in turn regulate HIF-1α.46,49
Targeting EGFR signaling for glioma therapy
EGFR signaling has been classically the central target for GBM therapy. The past 3 to 4 decades has seen the development of several strategies to curb EGFR activities ranging from small molecule inhibitors to engineered antibodies and viruses. Blocking the tyrosine kinase activity of EGFR has been the most attractive strategy in antitumor therapy. One approach has been to use several competitive and noncompetitive kinase inhibitors like erlotinib, gefitinib, and other second-generation inhibitors like afatinib, dacomitinib, often in combination with radiation and temozolomide. Another approach in targeting EGFR is using monoclonal antibodies (mAbs), like cetuximab, nimotuzumab, and so on, which have also shown preliminary antitumor effects in GBM. Apart from EGFR itself, various downstream effectors of the cascade have also been targeted with varying degrees of success (see Table 2 and references therein).
Pharmacological Compounds Targeting EGFR and Wnt/β-Catenin Pathways in Glioma.

Wnt Signaling
Canonical Wnt signaling (β-catenin dependent) regulates a wide set of genes that in turn orchestrate diverse cellular functions such as morphogenesis, differentiation, and proliferation. Consequently, aberrant activation of the canonical Wnt pathway has been found to be the driver of several human cancers including glioma. In the absence of Wnt ligands, the cell gets rid of excess β-catenin by assembling a multiprotein destruction complex composed mainly of Axin, APC (adenomatous polyposis coli), and GSK3β (glycogen synthase kinase 3-β), on which β-catenin is first phosphorylated by CK1α (casein kinase 1-α), followed by GSK3β and then ubiquitinated by the E3 ligase β-TrCP (β-transducin repeat containing protein) to be subsequently degraded through the 26S proteasome system. In the absence of nuclear β-catenin, the Tcf/Lef (T-cell specific transcription factor/lymphoid enhancer-binding factor) family of transcription factors (Tcf1, Tcf3, Tcf4, and Lef1) recruit the transcriptional co-repressors Groucho/TLE and efficiently repress expression of their target genes. The interaction of Wnt with Fzd/LRP (Frizzled/lipoprotein-receptor related protein) changes the state of things in ways for which a number of biochemical and cellular mechanisms have been proposed: first, it may trigger the formation of Dishevelled (Dvl)–Fzd complex, which in turn, sequesters Axin (and possibly GSK3β) to the plasma membrane thus blocking the formation of destruction complex85-90; second, Wnt induced phosphorylation of LRP6 at PPPSPXS motifs by GSK3β and CK1γ may create docking sites for Axin, facilitating its sequestration on the membrane and inactivation of the destruction complex91,92; third, Wnt induced phosphorylation of PPPSPXS motifs of LRP6 may inactivate the catalytic pocket of GSK3β, thus directly blocking its activity against β-catenin93-96; fourth, Wnt signaling may promote internalization of GSK3β by multivesicular bodies (MVBs), thus preventing it from phosphorylating β-catenin. 97 Although, all these models are relatively new and require further consolidation, it is conceivable based on the evidences that most of them are operational and may even complement each other. Nevertheless, the final outcome of these cascades is the stabilization of β-catenin, the central player of the pathway, which eventually accumulates inside the nucleus in a Rac-1 dependent manner 98 and serves as a transcriptional coactivator to Tcf/Lef99-101 (see Fig. 1).
β-Catenin is not a direct target for alterations but is a central player in glioma
The Wnt pathway was first linked to cancer when the APC gene was found to be mutated in both familial adenomatous polyposis coli (FAP) and sporadic colon cancers. Approximately 90% of sporadic colon cancers harbor mutations in APC (80%), with β-catenin and Axin 2 leading to chronic activation of Wnt signaling. Early evidences for the involvement of Wnt signaling in brain tumors came when similar germline mutations of APC were found in gliomas and later medulloblastomas.102,103 However, unlike in medulloblastomas where more than 30% of the cases carry an activating mutation in β-catenin gene (CTNNB1), these mutations per se are not so prevalent in GBM, apparently pointing to other mechanisms of β-catenin overactivation. Further support for an important role of β-catenin in glioma was provided by the observations that experimentally induced gliomas in rat using N-ethyl-N-nitrosourea display aberrant nuclear accumulation of β-catenin in contrast to normal brains.104,105 Proliferation of several glioma cells could be significantly inhibited by targeting the pathway using Wnt2 and β-catenin siRNAs104,106 or all-trans retinoic acid (ATRA). 107 ATRA is a potent differentiating agent, an activity that has been exploited for antitumor therapy by eradication of CSCs. 108 In glioma cells (U251 and C6) ATRA has been shown, at least in part, to act by upregulating Axin expression and cytoplasmic retention of β-catenin, thus trans-repressing Wnt/β-catenin signaling. 107 Increased β-catenin expression has been consistently found in astrocytic tumors relative to normal brain regions and has been correlated with poor prognosis and short survival of GBM patients.109,110
Recently, an interesting and fundamental mechanism for Wnt/β-catenin signaling activation in GBM was discovered involving the forkhead box (Fox) transcription factor, FoxM1, which is a well-known proto-oncogene in multiple cancers. In glioma, FoxM1 was found to be indispensable for nuclear localization of β-catenin because its nuclear translocation fails in FoxM1−/− cells. 111 An extensive crosstalk is now known to exist between Wnt/β-catenin and FoxM1, and the interdependency is such that depleting either protein functionally handicaps the other, alleviating the self-renewal capability of GSCs, induces differentiation, and inhibits glioma progression. 112
Inactivation of key components of the β-catenin degradation complex is common in brain tumors
APC, Axin, and GSK3β are the central molecules that together assemble the destruction complex on which β-catenin is phosphorylated and degraded through the ubiquitin-proteasome pathway. Apart from β-catenin, aberrations of these players have been documented in various forms of brain tumors. For example, although medulloblastomas appear sporadically, patients with FAP or Turcot’s syndrome are significantly at a higher risk of developing the malignancy. Such patients have been found to carry inactivating aberrations of APC (point mutation), Axin1 (deletion), and Axin2 (truncation) genes.102,103,113-115 As compared to normal brain ~30% of GBM tissues were found to contain lower levels of Axin1, and 69% of these samples contained predominantly cytoplasmic Axin1, which is inhibitory to Wnt/β-catenin signaling. 116 Furthermore, the levels of Axin significantly correlates negatively with the grades of astrocytoma, and its overexpression in C6 cells induced apoptosis and reduced proliferation by upregulating the expression of p53. 117
Inhibition of GSK3β by LiCl elicits morphological changes in U87MG cells and induces their proliferation. 118 In addition, GSK3β activation induces differentiation of human GBM cells, characterized by morphological and biochemical changes to the astrocytic phenotype, and decrease in proliferation. 119 Interestingly, observations against a tumor suppressive role for GSK3β have also been reported, which is in complete contrast to the aforementioned studies. For example, GSK3β inhibition (by siRNA, SB216763, or LiCl) have been shown to induce tumor cell differentiation and apoptosis.120,121 This apparent paradox has been explained by the fact that several growth-promoting pathways, such as EGFR, are commonly overexpressed in GBM. Alternatively, because cancer cells are low in differentiation and apoptosis inducing program, the effect of GSK3β inhibition is directly detectable. 121
Another positive regulator of Wnt/β-catenin signaling is FRAT1 (frequently rearranged in advanced T-cell lymphomas 1), which is emerging as an important factor in glioma tumorigenesis and progression. 122 FRAT1 belongs to GSK3β-binding family of proteins and inhibits GSK3β-mediated phosphorylation of β-catenin.123,124 The expression levels of FRAT1 is dramatically increased in gliomas and directly correlates with the proliferative capacity of glioma cells. Knockdown of FRAT1 in U251 GBM cells suppressed cell proliferation, migration, invasion in vitro, and significantly impaired tumor xenograft growth in nude mice.125,126
Promoter hypermethylation of Wnt antagonists is a frequent alteration in glioma
The hallmark of activated Wnt signaling is the preferential nuclear accumulation of β-catenin. In cancer, naturally occurring mutations in various components of the pathway has achieved this by multiple means. Mutations of APC and Axin inhibit formation of the β-catenin destruction complex and those of β-catenin at conserved phosphorylation sites blocks its degradation, both leading to aberrant stabilization and nuclear accumulation of β-catenin. In addition to these key culprits, mutations in other components of the pathway, such as Tcf4, ICAT (inhibitor of β-catenin and Tcf4), PP2A (protein phosphatase 2A), and GSK3β, have also been reported to overactivate Wnt signaling. These are prevalent in cancers of the colon, liver, ovary, skin, prostate, melanoma, and endometroid ovarian cancer.127,128 In addition to these mutations, many cancers carry silenced (epigenetically inactivated) genes encoding family of Wnt antagonists such as sFRP (secreted Fzd-related protein) and WIF1 (Wnt inhibitory factor 1), which act as natural brakes of the Wnt pathway by binding Wnt ligands to block activation at the cell surface. Malignancies of the human brain (gliomas and medulloblastomas) seem to have preferably taken this latter approach. Large-scale epigenomic analysis has shown the promoters of Dkk1 (dickkopf 1 homolog), sFRP1, and WIF1 to be heavily methylated in GBM and other astrocytomas,129,130 and their relative expressions were found to directly correlate with tumor grades. At least one of these genes were found to be hypermethylated in 6 of 16 diffuse astrocytomas (38%), 4 of 14 anaplastic astrocytomas (29%), 7 of 10 secondary glioblastomas (70%), and 23 of 30 primary glioblastomas (77%). Hypermethylation of sFRP1, sFRP2, and Nkd2 (naked 2) each occurred in more than 40% of the primary glioblastomas, while Dkk1 hypermethylation was found in 50% of secondary glioblastomas. Deletions of WIF1 gene has also been observed in approximately 10% of gliomas. Importantly, these studies also demonstrated that ectopic expression of these silenced genes could alleviate tumor-associated characteristics such as colony formation and tumorigenesis in vivo as well as induce apoptosis.131,132
Noncanonical Wnt signaling in glioma
The noncanonical Wnt signaling (β-catenin independent) has been primarily implicated in determining the cellular polarity, ciliogenesis, calcium signaling, cardiogenesis, and modulating the activity of a histone lysine methyltransferase through the CaMKII-NLK (Ca2+/calmodulin-dependent protein kinase II–Nemo like kinase) axis.133-137 Wnt5a, a classical component of noncanonical Wnt signaling, is the most predominant and commonly overexpressed member of the Wnt family in glioma cell lines and tissues. 138 Overexpression of Wnt5a could increase, while downregulation could reduce, proliferation and tumorigenicity of GBM-05 and U87MG cells. Knock-down of Wnt 5a suppresses migration and infiltrative capacity of glioma cells partly in a MMP2 dependent manner.139,140 In primary GBM cells, depletion of Wnt5a causes induction of senescence and reduction of neurosphere-forming capability. 141 Depletion of Dvl2, a crucial component of both canonical and noncanonical Wnt signaling, severely retards proliferative capacity of glioma cells both in vitro and in vivo, via the noncanonical path. 141 Furthermore, Wnt driven planar cell polarity (PCP) signaling suppresses endothelial cell proliferation and migration, supporting tumor promoting function of β-catenin independent Wnt pathways. 142 Recently, it has been shown that the Wnt-specific secretory protein Evi (also known as GPR177/Wntless/Sprinter), a core Wnt signaling component affecting both canonical and noncanonical signaling, is significantly overexpressed in astrocytic gliomas and its depletion in glioma cells (U87MG, A172, U251MG, and LN229) and glioma-derived GSCs led to decreased cell proliferation and apoptosis, in vivo tumor growth, and migration effectuated by strong reduction in the expression profiles of several pro-oncogenic transcription factors and interleukins. 143 Evidences are rapidly growing to support an important role for noncanonical Wnt signaling in glioma; however, for the purpose of this review we generally limit ourselves to the canonical Wnt signaling as mediated through β-catenin.
Wnt is a major pathway involved in sustenance of GSCs
The presence of CSCs in most of human solid tumors has attracted tremendous interest in pathways important for the propagation and maintenance of these cells. The proof-of-concept that these pathways can indeed represent highly potential therapeutic targets are derived from experiments showing, for example, conditional deletion of β-catenin can result in complete regression of the CD34+ CSC xenograft tumor model of malignant human squamous cell carcinomas. 144 Similarly in GBM a subpopulation of cells identified by the expression of CD133 marker were isolated and characterized to be GSCs.145,146 Although, later studies have questioned the authenticity of CD133 as a bona fide marker of GSCs and instead have forwarded a number of other markers such as CD15, CD44, ID1, L1CAM (L1 cell adhesion molecule), Integrin α6 among others, the existence of GSCs itself in glioblastoma is beyond doubt. 147 Knowledge from other cancers has shown that these cells are responsible for the aggressiveness, refractoriness-to-therapy, and relapse, characteristic to each tumor type. Molecular deciphering of each tumor type and more specifically the CSC population harbored within is of paramount importance to be able to target these cells. The potential therapeutic opportunity of GSCs is now widely accepted and is an area of active research.148,149
The definitive role of the Wnt/β-catenin pathway in regulating various aspects of stem cells is now well accepted and therefore has gained much attention from a therapeutic perspective.150,151 Wnt pathway is essential for maintaining the niche of NSCs such that its inhibition and overactivation has a direct influence on cycling and expansion of NPCs. 152 Furthermore, Wnt pathway genes have been shown to drive the malignant transformation of non-tumorigenic primary human mammary epithelial cells to mammary stem cells by regulating the DNA damage response at multiple levels to promote chromosomal instability and prevent mitotic defects and apoptosis. 153 Positive Wnt signaling regulators such as Fzd 4 are upregulated in a manner related to the invasiveness of the GSCs and correlates with resistance to anticancer drugs, neurosphere formation, and epithelial-to-mesenchymal transition (EMT). 154 Because pathways regulating stem cell renewal and differentiation are intimately coupled, it can be logically assumed that other potentially important and yet unidentified factors must be involved. Indeed, one such factor was recently shown to be PlagL2 (pleiomorphic adenoma-like gene 2). This gene is frequently targeted for copy number gain/amplification in human malignant gliomas and colon cancers. PlagL2 transcriptionally upregulates Wnt ligands (Wnt6) and its cognate receptors (Fzd9 and Fzd2), thus stabilizing β-catenin and allowing the GSCs to assume indefinite proliferative capacity. Furthermore, PlagL2-mediated suppression of NSC differentiation was partially dependent on Wnt signaling because attenuation of Wnt signaling in PlagL2-expressing NSCs through either Dkk1 overexpression or Wnt6/Fzd2 knockdown reverses PlagL2-mediated differentiation-suppression activity. 155
Targeting canonical Wnt signaling for glioma therapy
Wnt signaling presents several avenues for antitumor therapy. Possibilities include targeting receptors, proteases, kinases, use of small molecules to target β-catenin, or the other players involved in Wnt signaling cascade. Being the central player in the canconical Wnt pathway, blocking either β-catenin interaction with Tcf/Lef to block its transcriptional activity or its interaction with other protein (interacting partners) is widely exploited. 156 Engineered viruses had been developed as a tool in the last decade to target Wnt signaling—lytic viruses that could replicate only in cells with aberrant β-catenin signaling, viral antigens controlled by Tcf4 promoter, 157 inserting transgenes into viral genome to target drugs only to the infected cells.158,159 Some of the other approaches were small molecule inhibitors against Wnt/β-catenin signaling, 160 RNAi-mediated silencing of β-catenin,161-163 or even repression of β-catenin transcription complex, for example, by binding to orphan nuclear receptor ROR-α 164 (see Table 2).
Interactions between EGFR and Wnt/β-catenin pathways are complex and occur at multiple levels
Cross-communication between various signaling pathways allows the integration of a myriad of external and internal stimuli, the net outcome of which in cancer cells is unabated proliferation. EGFR and Wnt/β-catenin pathways are known to interact under malignant conditions, but how these extensive collusions occur, and the potential ramifications, remains an important and interesting topic (see Fig. 2).
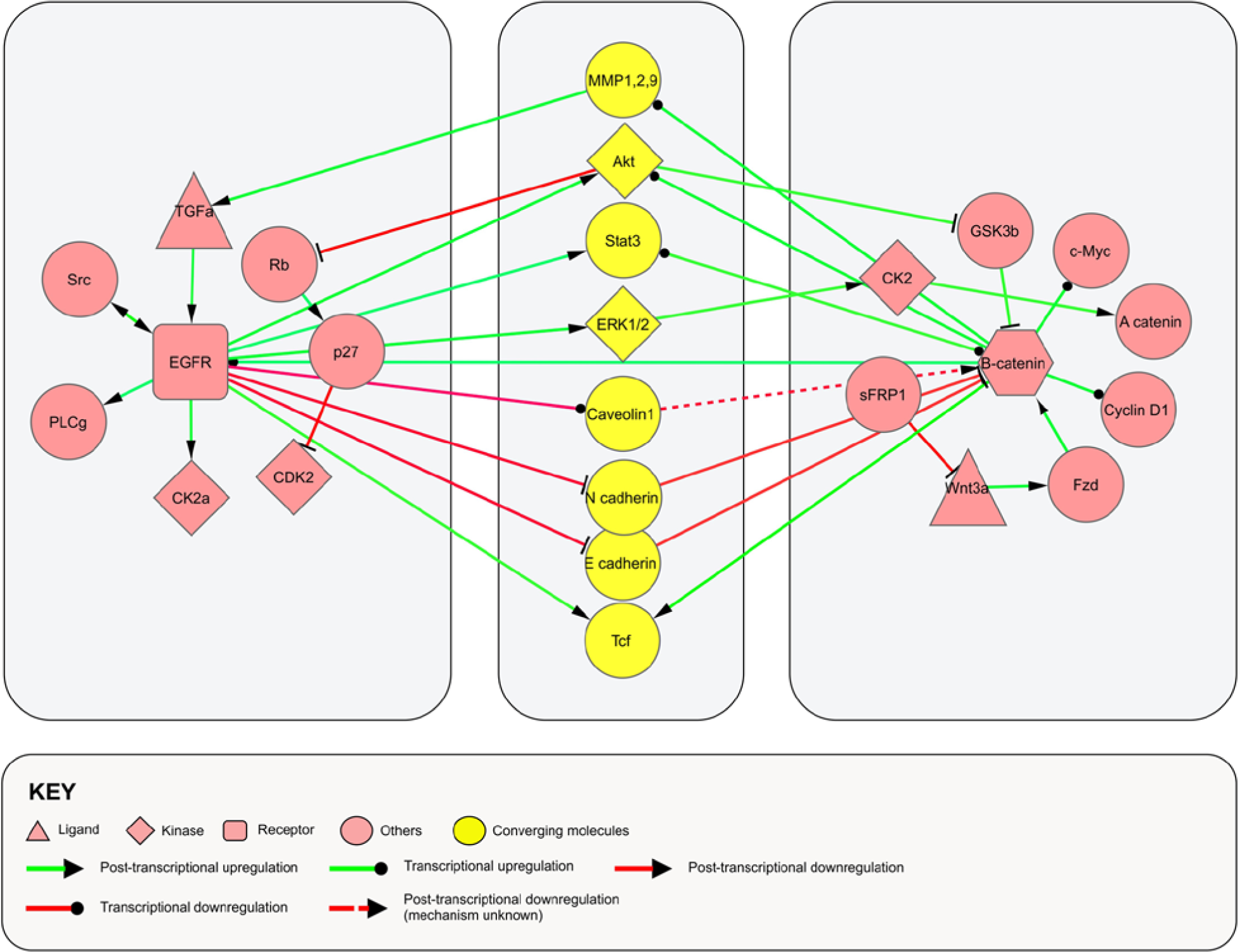
Schematic representation showing points of convergence between EGFR and Wnt/β-catenin pathways. The major players of EGFR and Wnt/β-catenin pathways are shown in
Transcriptional
Intratumoral administration of β-catenin siRNA into subcutaneous LN229 gliomas in nude mice has been shown to transcriptionally downregulate EGFR, STAT3, Akt1, Cyclin D1, MMP2, and MMP9. 165 Reciprocally, transcriptional activity of β-catenin has been found to be significantly correlated to Akt1 expression in glioma cells.32,111,165,166 In breast cancer cell lines (MCF7 and BT-474), STAT3 upregulates the protein level of β-catenin, by binding at 2 STAT3-binding sites on β-catenin promoter, and enhances its transcriptional activity. 167 Analysis of liver isolated from transgenic mice overexpressing wild-type β-catenin revealed increased levels of EGFR and STAT3 mRNA and protein together with their activated forms. 168 Increased EGFR promoter activity were observed on Wnt3a treatment, presumably through a Tcf binding site on EGFR promoter, which was abrogated by sFRP1. 168 Similar activation of EGFR promoter through conserved Tcf binding elements was observed on LiCl treatment, which was abrogated on destruction of those sites. 169 Apart from a direct reciprocal transcriptional regulation between EGFR and Wnt/β-catenin signaling, both these pathways activate and/or repress genes that are in turn crucial regulators of each other. For example, EGFR activation results in Wnt-independent β-catenin transactivation by transcriptional downregulation of caveolin-1. Overexpression of caveolin-1 suppresses the effect of EGFR on β-catenin activity. Caveolin-1 inhibits Wnt/β-catenin signaling by sequestering β-catenin to caveolae membrane domains. 170 Yet another instance of Wnt-independent transactivation of β-catenin was shown in multiple human cancer cell lines where activated EGFR induced nuclear translocation and binding of PKM2 (pyruvate kinase muscle isozyme) to phospho-β-catenin Y333 (c-Src-mediated phosphorylation), leading to deacetylation of the promoter and expression of Cyclin D1 and c-Myc. A positive correlation in the levels of phospho-β-catenin and nuclear PKM2 with the various grades of glioma malignancy and prognosis was also reported showing that β-catenin was responsible for activated EGFR-induced tumorigenesis and tumor cell proliferation. 171
Posttranslational
Wnt-independent and EGFR-dependent activation of the β-catenin pathway has been observed in the context of UVA irradiation in keratinocytes. Phosphorylation of β-catenin at Y654 by EGFR leads to its dissociation from E-cadherin/α-catenin membrane pool and consequent nuclear accumulation. Inside the nucleus, β-catenin/Tcf4 complex upregulates MMP1 leading to loss of cell-to-cell contact and increased cell migration. 172 EGFR also induces β-catenin-mediated transcription via an EGFR-Erk-CK2 axis. Treatment of EGF in EGFR overexpressing U87E, U373, and LN229 human glioblastoma cells and A431 human epidermoid carcinoma cell activates Erk2, which in turn directly binds to CK2α via the Erk2 docking groove and phosphorylates CK2α at T360/S362, thus activating it. CK2-mediated phosphorylation of α-catenin at S641 in turn abrogates the inhibitory effect of α-catenin on β-catenin. Phosphorylation levels of α-catenin at S641 correlates with the level of Erk1/2 activity in human glioblastoma specimens and with the grades of glioma malignancy. 173
Another point of intersection is through Akt, which is an activated downstream member of EGFR signaling. In A431, CHOAA8, and HEK293T cells, Akt-mediated phosphorylation of β-catenin at S552 was found to cause its dissociation from the E-cadherin pool, leading to β-catenin accumulation in both the cytoplasm and the nucleus that enhances its interaction with 14-3-3ζ via a binding motif containing phospho-S552 and increased transcriptional activity that promotes tumor cell invasion and development. 174 S552 phosphorylation can also be mediated by PKA. Again, phospho-S675 β-catenin (by PKA) interacts with CBP (CREB-binding protein), which illustrates that PKA promotes transcriptional activity of β-catenin. A recent study has shown that Src-mediated Y654 phosphorylation of β-catenin facilitates the PKA-mediated phosphorylation at S675, thus enhancing Wnt signaling mediated intestinal tumor initiation in APC deficient mice.175,176 These findings highlight the importance of the crosstalk between EGFR and Wnt pathways in tumor development.
Evidences for a functional interaction between EGFR and canonical Wnt pathways
A number of reports have shown EGFR and Wnt pathways extensively collaborate in neoplasias and that this collaboration is important for maintaining the neoplastic state. The observation that EGFR mutations were associated with a relatively better prognosis in non–small cell lung cancer patients with unmethylated Wnt antagonists than in patients with methylations clearly points to a functional cooperation. 177 Apart from such patient-derived observations, reports from in vitro studies also strengthen this notion. In multiple oral cancer cells, activated EGFR promotes angiogenesis and cell migration by upregulating VEGF and MMPs, an effect at least partially dependent on induction of β-catenin nuclear translocation. 178 Reciprocally, activation of β-catenin by inactivation of E-cadherin activates EGFR, thus effectively creating a positive feedback loop. Inhibition of EGFR in LN18 glioma cells by either using inhibitors or overexpressing dominant negative receptor isoforms (HER2VEKA) can interfere with migration. The nonmigratory phenotype is mediated through upregulation of N-cadherin and its recruitment to the cell membrane. Interestingly, downregulation of N-cadherin is accompanied by a direct interaction of EGFR-HER2 heterodimers with N-cadherin–β-catenin complexes, leading to tyrosine phosphorylation of β-catenin. 179 Silencing of EGFRvIII reduced the expression of factors involved in EMT including N-cadherin, β-catenin, Snail, Slug, and paxillin, with a decrease in cell cycle progression in U87MG cells. 180 Constitutive activation of PDK1, Akt, and phosphorylation of GSK3β is a common feature of pediatric gliomas. Small-molecule inhibitors of PI3K/Akt signaling pathway such as OSU03012 activate GSK3β, resulting in degradation of β-catenin and reduced expression of its target genes Cyclin D1 and c-Myc. 181 β-Catenin is overexpressed in human glioblastoma and knockdown of β-catenin inhibits glioblastoma cell proliferation and invasive ability, and induces apoptotic cell death. Furthermore, intratumoral introduction of siRNA targeting β-catenin into established subcutaneous gliomas also delayed the tumor growth. Both in vitro and in vivo studies have confirmed that downregulation of β-catenin leads to reduced expression of EGFR, STAT3, Cyclin D1, MMP2, MMP9, and Akt1 mRNA as well as protein with a concomitant decrease of their active forms. All these observations clearly point to functional cooperation between EGFR and canonical Wnt pathways.77,105,106,109,111,166
Another not yet explored point of collusion between these 2 pathways is microRNAs (miRs). For example, miR-21 is an important miR in gliomagenesis and has been shown to upregulate the EGFR pathway through Akt by downregulating PTEN. miR-21 also upregulates STAT3. Both Akt and STAT3 are reported to activate β-catenin signaling. miR-21 itself is under transcriptional regulation of FOXO3a, STAT3, and activator protein-α.182-184 Thus, further research would enable a comprehensive understanding of the miR network, controlling both EGFR and canonical Wnt pathways in glioma.
Cooperation of EGFR and Wnt pathways in GSCs
With the ever-increasing appreciation regarding the centrality of GSCs in glioma pathology and their establishment as valid therapeutic targets, controlling the proliferation of this subpopulation of cells is now seen by many as a priority.148,149 Complex signaling crosstalks between multiple extracellular ligand-induced pathways regulate the maintenance and proliferation of these cells. Both EGFR and Wnt pathways regulate multiple aspects of NSCs like migration, proliferation, and survival.41,151 Because Wnt and EGFR signaling have been shown to complement each other in various contexts, it will be logical to assume that they do so in the case of GSCs as well.
A point to remember is that Wnt and EGFR pathways are distinct cascades and provide quite different set of instructions to NSCs. However, extensive genetic reprogramming may result in aberrant and ectopic activation and as a result bring the tumorigenic potential of these pathways into reinforcing each other in driving malignancies. As, for instance, overexpression of oncogenic H-Ras (L61) has the ability to transform p53-deficient oligodendrocyte precursor cells (OPCs) and NPCs into GSCs in mice. As few as 10 GSC-like cells when transplanted in vivo form a retransplantable GBM. GSCs obtained from such tumors could be killed using a combination of PTGS2/COX2 and EGFR-inhibitor. 184 Overactivation of both EGFR and Wnt signaling pathways has been documented in GSCs and is thought to be responsible for refractoriness to current therapies.48,185 Coordination between STAT3 signaling and Wnt5a expression has been observed in glioblastoma. Activated STAT3 pathway may induce transcriptional upregulation of Wnt5a, whereas Wnt5a knockdown suppressed STAT3 regulated genes.186,187 Although detailed study asking what roles these different pathways may play to complement or supplement each other in the maintenance of GSCs is absent, evidences exist to indicate toward such a possibility. For instance, cultured U87MG and T98G GBM cells in the presence of EGF and bFGF can be induced to form gliospheres (glioma sphere) that constitute more than 50% CD133+ GSCs. Such gliosphere formation can be inhibited by treatment with peroxisome proliferator activated receptor γ (PPARγ) agonist, 15-Deoxy-Delta(12,14)-Prostaglandin J(2) (15d-PGJ2), or ATRA, in a reversible and dose-dependent manner with concomitant cell cycle arrest and induction of apoptosis. 188 PPARγ agonists pioglitazone, ciglitazone, and rezulin significantly inhibit glioma cell proliferation and induce apoptosis.189-191 In colon cancer, aberrant Wnt/β-catenin signaling causes nuclear accumulation of PPARγ, facilitating tumor progression. 192 Recently, induction of PPARγ expression was observed on EGFR inhibition. In lung and bladder cancers, combination of PPARγ agonists and EGFR antagonists displayed better efficacy for antitumor therapy.193,194 Thus, the outcome of effects of PPARγ on Wnt/β-catenin signaling and EGFR signaling and vice versa needs to be adequately investigated, which represents a potential point of crosstalk. These observations, therefore, may support the notion that EGFR and Wnt pathways collaborate in GSC maintenance.
Concluding Remarks and Future Directions
Signaling pathways do not act in isolation and multiple signaling modules collaborate to elicit responses at the cellular level. For example, complete blockade of EGFR signaling does not result in apoptosis of human glioma cells, suggesting involvement of additional pathways. Also, regulation of downstream signal transducers in the EGFR pathway seems to be dominated by regulatory circuits independent of EGFR phosphorylation. 195 Therefore, newer approaches targeting multiple collaborating pathways regulating varied aspects of a tumor demands active attention. For example, along with classical cytotoxicity-based drugs administered for their cell killing activity, drugs targeting specifically CSCs can be combined to achieve significant effectiveness in eradicating tumors.196,197
The convergence of EGFR and Wnt/β-catenin pathways is apparent over a wide range of molecular crosstalks giving rise to differential outcomes. 198 In the perspective of this review, we have projected several instances taken as much as possible from glioma to highlight the interdependency of these 2 pathways toward malignant development collectively contributing to the poor prognosis and refractoriness to therapy. Targeting both the pathways using specific drugs, mAbs, inhibitors, RNAis, or engineered viruses simultaneously could prove to be beneficial. Use of certain drugs, mostly from natural sources like quercetin and curcumin, has shown some initial promise (see Table 2). The GSCs are a potential point of convergence of these signaling, and in this combinatorial approach, common factors like hypoxia, CD44 and/or CD133 markers, stem cell niche, tumor microenvironment, VEGF, and so on, need to be exploited. As small molecule inhibitors targeting these signaling pathways continue to be developed, highly representative model systems of human GBM (both genetic and xenograft) will be necessary to provide a reasonable picture of the targetable molecular phenotype of GBM. For example, consistently expressing EGFR and EGFRvIII in both subcutaneous xenografts and in culture under stem cell conditions have been used to study the effect of inhibitors targeting various aspects of EGFR and Wnt signaling at multiple points. 199 In an interesting finding employing murine glioma model and GL261 glioma cells, Reis and coworkers reported that sustained endothelial Wnt/β-catenin signaling actually results in vascular quiescence and diminished angiogenesis leading to shunted tumor growth. 138 Clearly, the Wnt/β-catenin pathway is an interesting target that could be exploited for GBM therapy. Therefore, a proper understanding of the contributions of EGFR and Wnt/β-catenin signaling in regulating various anatomical aspects of glioma is necessary.
In a conceptual model (Fig. 3), we speculate the possible combinatorial approaches, by targeting multiple pathways together in the context of signaling crosstalk. Because a tumor has multiple cellular elements that interact with each other in complex ways to generate the enormous diversity at the molecular level, a universal cure for cancer may not be a plausible proposition. However, a unifying theme has emerged. First, all cancers need to acquire the hallmark capabilities, for which they are totally dependent on critical growth factor signaling (e.g., EGFR signaling). Second, CSCs represent the true epicenter of a tumor. These stem cells rely on defined signaling pathways (e.g., Wnt signaling) for their continued revival and timely differentiation. Third, stromal and tumor cells extensively communicate, which may be critical in maintaining certain types of tumors. The challenge is to fully understand these various processes that demand novel tools to enable multiparametric analysis encompassing dependencies of tumors on signaling networks and interdependencies between these networks. Gaining this knowledge would enable anticipation of outcomes and help direct effective combination treatments.
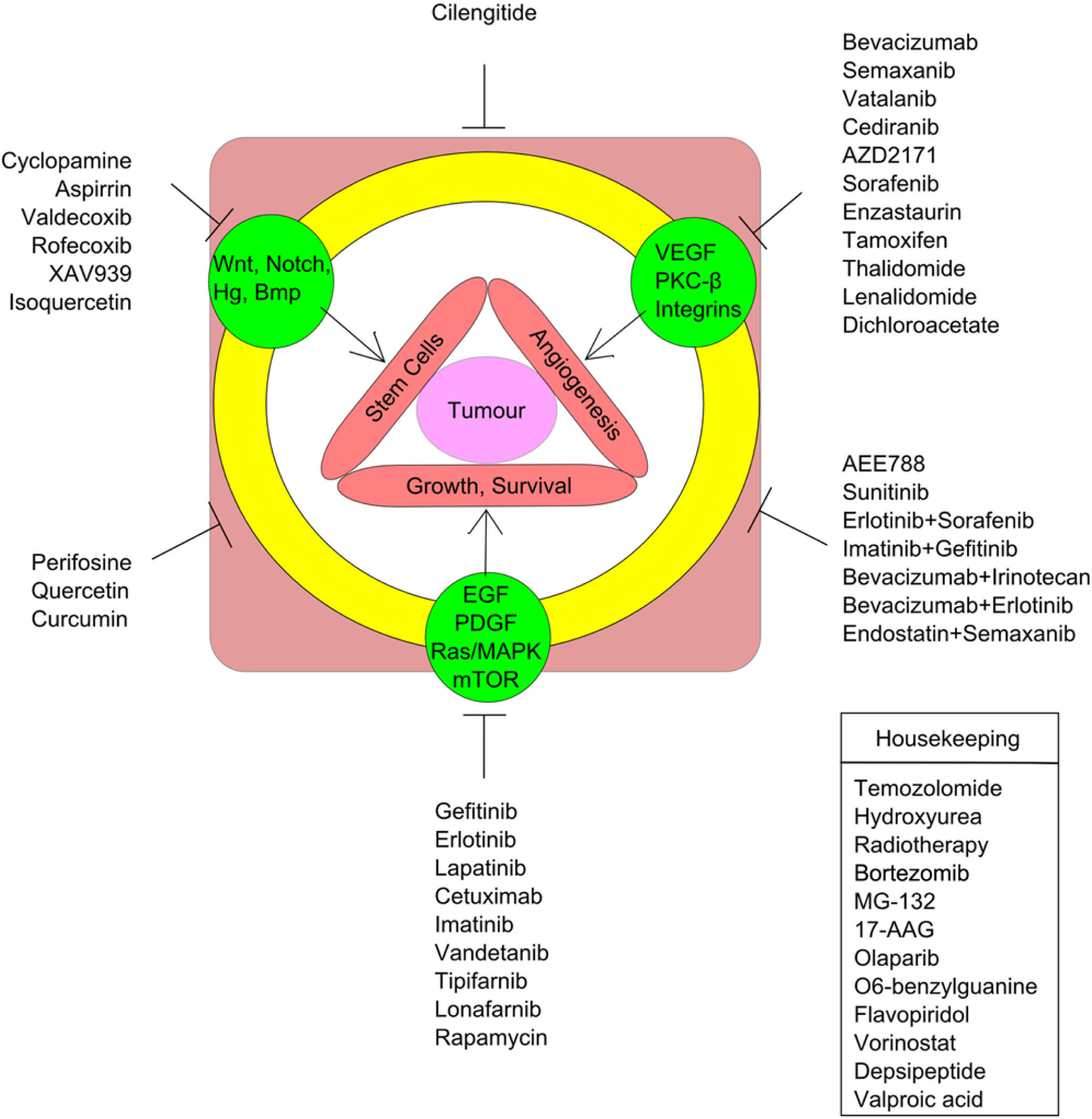
Schematic representation showing various signaling pathways regulating distinct aspects of a tumor and current status of their therapeutic exploitation. The figure also highlights windows that can be actively considered for future drug development. A tumor (central pink circle) can be thought of being therapeutically targeted at its 3 cardinal features: growth and survival, angiogenesis, and stem cells (red bars). Research, in turn, has shown that these 3 properties are being driven by certain major cellular signaling pathways (green closed circles), which have been the center of attraction for clinical intervention. All these signaling pathways exhibit extensive crosstalk (yellow open circle) and thus both these individual pathways (classically) and their interjunctions (combinatorial approach, more recently) have been used for drug development, as shown in the figure. For example, cilengitide has been shown to be both anti-angiogenic and also inhibits proliferation of glioma stem cells. The figure clearly shows that most drug development efforts have been concentrated toward the angiogenesis and growth/survival properties. However, drugs targeting angiogenesis and stem cells combinatorially are relatively unexplored. The inset box lists drugs that target generalized housekeeping properties of tumor cells in contrast to a specific signaling pathway.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Our laboratory is supported with grants provided by Council of Scientific and Industrial Research (EMPOWER [OLP2]; MEDCHEM [BSC0108]; Systems Biology [HCP004]) and the Department of Science & Technology (SR/SO/HS-150/2010), Government of India.