
Abstract
The FLT3 tyrosine kinase receptor is involved in both hematopoiesis and hematological malignancies. The Wnt/β-catenin pathway has been shown to participate in the self-renewal of hematopoietic stem cells and to cooperate with the mutant FLT3 receptors in leukemic transformation. However, the detailed biological impact of such a constitutively activated Wnt pathway remains to be further explored. Here, the authors report that activating mutations of FLT3 constitutively activate β-catenin by inhibition of GSK-3β in a PI3 kinase pathway–dependent manner. Ectopic expression of a dominant negative form of GSK-3β in FLT3-ITD–expressing cells activated β-catenin and blocked the downregulation of the TCF/β-catenin transcriptional activity induced by inhibition of FLT3 kinase. Furthermore, inhibition of cell proliferation and colony formation induced by such suppression of FLT3 kinase activity could be partially reversed by knockdown of GSK-3β and restored by knockdown of either TCF4 or β-catenin. Moreover, exogenous activation of the Wnt pathway also attenuated such inhibitory effect. These findings indicate that the potencies of the inhibitors of FLT3 kinase activity could be modulated by the activity of the Wnt/β-catenin pathway in the cells harboring FLT3-ITD mutations, and FLT3-ITDs signal through GSK-3β to activate β-catenin that this is likely to directly contribute to the leukemic phenotype.
Introduction
The FLT3 tyrosine kinase receptor plays a critical role in hematopoiesis 1 and is also involved in leukemogenesis. 2,3 Gain-of-function mutations of FLT3 have been found in approximately 30% to 35% of patients with acute myeloid leukemia (AML), most commonly as in-frame insertions of variable length (internal tandem duplications [ITDs]) in the juxtamembrane domain and the missense point mutations in the kinase domain. Although these mutations have transforming activity, 4,5 results from a murine bone marrow transplantation assay 6 suggested that FLT3-ITD mutations needed to cooperate with other oncogenes to induce AML. 7
The Wingless-type (Wnt) pathway has been shown to participate in self-renewal of hematopoietic stem cells 8 and to act synergistically with the FLT3-ITD in leukemic transformation. 9-11 Although activation of the Wnt/β-catenin pathway via direct interaction between the FLT3-ITD and β-catenin has been reported, 11 it has also been shown that in 32D-FLT3-ITD and 32D-cKit-ITD cells, the ITD mutant kinase activities were associated with GSK-3β and β-catenin activities, although the autocrine mechanism seemed to be ruled out in this system. 12 In the canonical Wnt pathway, the central player is β-catenin, whose activity is regulated by a cytosolic destruction complex containing AXIN, adenomatous polyposis coli, and GSK-3β as major components. 13,14 Stimulation of the Wnt ligand inhibits the destruction complex and activates β-catenin, which functions as a transcriptional coactivator for lymphoid enhancer–binding factor 1/T cell-specific transcription factor (TCF) 15-17 to activate its target genes, such as c-Myc and cyclin D1. 18,19
The β-catenin regulations by the destruction complex have been shown to be cell type specific. Phosphorylation of β-catenin by GSK-3β is a critical step in the regulation of the Wnt signaling. Inhibition of GSK-3β by either its dominant negative form or lithium resulted in activation of TCF target transcripts in C57MG fibroblast cells but not in T-lymphocytes. 20 Also, in NIH3T3 cells, inhibition of GSK-3β by AKT was not sufficient to induce TCF target genes. 21 Thus, regulation of β-catenin needs to be studied carefully in each model system separately.
In this report, we further study the interaction between the FLT3-ITD and the Wnt signaling and specifically investigate β-catenin activation and the resulting biological effects in the context of the gain-of-function mutations of FLT3. Our results demonstrated that FLT3-ITDs signal GSK-3β to activate β-catenin and its downstream targets in a PI3 kinase pathway–dependent manner and that activation of the Wnt/β-catenin pathway in the context of activating mutations of the FLT3 receptor could modulate the potencies of the FLT3 kinase inhibitors.
Results
The Importance of the PI3 Kinase Pathway on the Constitutive Inhibition of GSK-3β and Activation of β-catenin in the Context of Mutant FLT3 Receptors
The activating mutations of FLT3 could render the growth of the murine factor IL3 dependent Ba/F3 and 32D cells factor independence and constitutively activated many downstream signaling pathways, including the PI3K/AKT and p44/42 MAPK pathways. 22,23 Such IL3-independent growth biologically distinguishes the Ba/F3 cells expressing FLT3 activating mutations from the parental Ba/F3 cells expressing either empty vector or wild-type FLT3 that could not render the Ba/F3 cells IL3 independence. Because GSK-3β is a potential substrate of the PI3K/AKT pathway, 24 we first looked at the activity of GSK-3β in the Ba/F3 cells expressing known activating mutations of the FLT3 receptor, FLT3-ITD and FLT3-N841I, 5 as shown in Figure 1A. The phosphorylation of GSK-3β at serine 9, which inhibits the activity of GSK-3β, 24-26 was increased in the mutant receptor expressing cells (compare lanes 3 and 5 to lanes 1), suggesting that GSK-3β was inhibited by the mutant FLT3 receptors. More importantly, such reduced GSK-3β activity was restored (lanes 4 and 6 of the pSer9GSK3β blot) when the autophosphorylation of the mutant FLT3 receptors was inhibited by PKC412 (lanes 4 and 6 of pTyr blot), a small-molecule inhibitor of the FLT3 kinase activity, 27 indicating a dependence of such reduction on the mutant receptors.
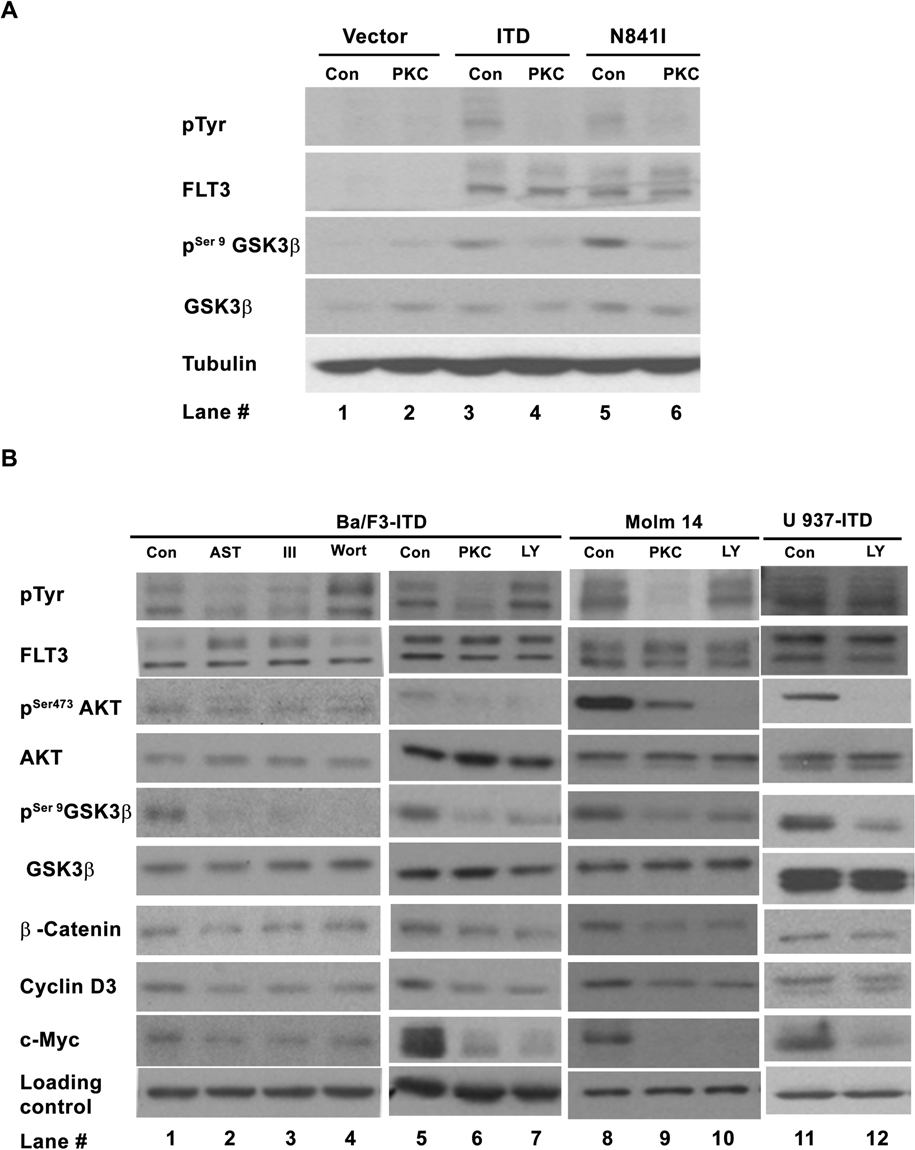
Importance of the PI3K/AKT/GSK-3β on the constitutive activation of β-catenin in mutant FLT3 receptor–expressing cells. (
GSK-3β activity is controlled by multiple signaling pathways 28,29 ; to determine the importance of PI3 kinase signaling, an inhibitor of the PI3 kinase pathway, LY294002, was used for further study. In the cells either ectopically expressing FLT3-ITD (Ba/F3-ITD and U937-ITD) or derived from an AML patient sample harboring FLT3-ITD (Molm 14; Fig. 1B, lanes 7, 10, and 12), LY294002 reduced the level of phosphorylation at serine 473 of AKT (pSer473AKT) and resulted in decreased levels of phosphorylation at serine 9 of GSK-3β (pSer9GSK-3β), as well as decreased β-catenin, c-Myc, and cyclin D3. These results were further confirmed by a structurally unrelated PI3 kinase inhibitor, Wortmannin, in Ba/F3-ITD cells (Fig. 1B, lane 4). These 2 inhibitors had not much effect on the tyrosine phosphorylation of the mutant FLT3-ITD receptor (pTyr blot of Fig. 1B, lanes 4 and 7; Supp. Fig. S1, lanes 4 and 7). By contrast, an inhibitor of the p44/42 MAPK pathway, U01126, had no effect on the level of β-catenin (data not shown). These data demonstrate that in the absence of IL3, activation of the PI3 kinase pathway results in activation of AKT, inhibition of GSK-3β kinase activity, and increased levels of β-catenin, c-Myc, and cyclin D3 in the FLT3-ITD–expressing cells. Furthermore, 3 FLT3 kinase inhibitors, PKC412, AST 487, 30 and inhibitor III, 31 reduced the level of tyrosine phosphorylation of the mutant FLT3 receptor (pTyr blot of Fig. 1B, lanes 2, 3, and 6; Supp. Fig. S1, lanes 2, 3, and 6), as well as reduced the levels of pSer473AKT, pSer9GSK-3β, β-catenin, c-Myc, and cyclin D3 (Fig. 1B, lanes 2, 3, 6, and 9), suggesting the dependence of the activations of PI3K/AKT/GSK-3β and β-catenin on FLT3-ITD. Although we do not rule out other potential signaling pathway(s) that may also regulate β-catenin activity, our results strongly indicate that the PI3 kinase pathway, most likely via the AKT/GSK-3β axis, is important for the activation of β-catenin in the context of FLT3-ITD.
FLT3-ITD Constitutively Activates β-catenin via Inhibition of GSK-3β
Although GSK-3β is an important component in the β-catenin destruction box, its regulatory capacity is cell type specific. To determine such capacity in the context of gain-of-function mutations of FLT3 receptor, we investigated the effects of GSK-3β inhibition on the activity of β-catenin.
Pharmacologic Inhibitions of GSK-3β Increase Levels of β-catenin, c-Myc, and Cyclin D3 and Enhance TCF/β-catenin Transcriptional Activity
Two commonly used inhibitors of GSK-3β, lithium chloride 32,33 and inhibitor VII, were first tested as shown in Fig. 2A (lanes 2, 3, 5, 6, 8, and 10). Both inhibitors suppressed the kinase activity of GSK-3β, as detected by the increased level of its phosphorylation at serine 9, and had not much effect on the tyrosine phosphorylation on the mutant FLT3-ITD receptor (pTyr blot of Fig. 2A, lanes 2, 3, 5, 6, 8, and 10; Supp. Fig. S1, lanes 10 and 11). The level of β-catenin went up in response to both drugs, indicating that inhibition of GSK-3β alone resulted in accumulation of β-catenin. We then used c-Myc and cyclin D3 to monitor β-catenin activity. In both Ba/F3-ITD and Molm 14 cells, their levels went up upon inhibition of GSK-3β associated with accumulation of β-catenin. In U937-ITD cells, the level of cyclin D3 (lane 10) went up following lithium treatment; the level of c-Myc did not change. It is possible that in U937 cells, expression of FLT3-ITD already resulted in a maximum accumulation of c-Myc (Fig. 2A, compare lane 9 with lane 7). These results indicate that pharmacologic inhibition of GSK-3β in the FLT3-ITD–expressing cells is able to activate β-catenin.
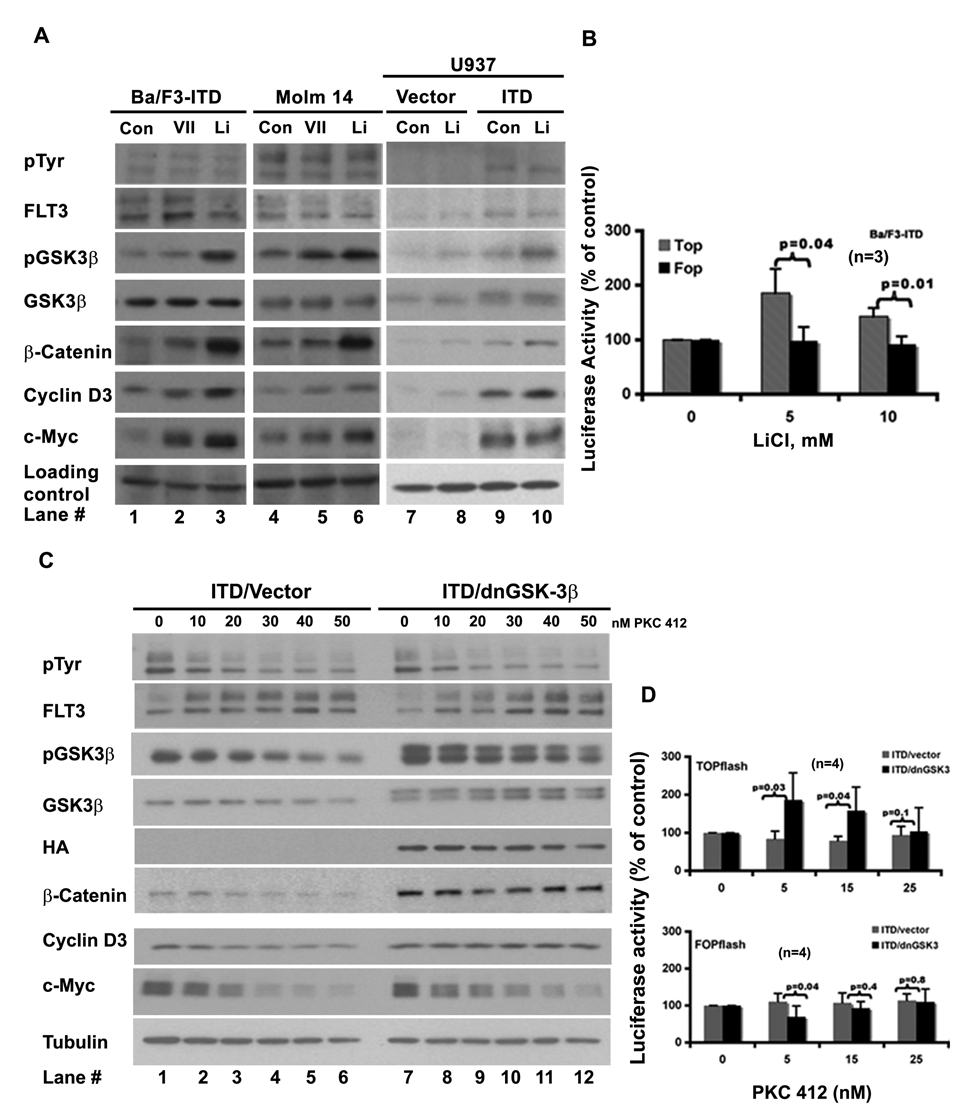
Inhibitions of GSK-3β activate β-catenin and attenuate the regulation of the mutant FLT3 receptor on the β-catenin activity. (
We then looked at the effect of LiCl on the TCF/ β-catenin driven promoter activity using the TOPflash/FOPflash TCF reporter system. In the TOPflash reporter, a full-length ORF of luciferase is driven by a TK minimal promoter with TCF binding sites, whereas in the FOPflash reporter, the TCF binding sites were mutated. Figure 2B shows that at the tested concentrations, LiCl was able to induce the activity of TOPflash (gray/stripe bar). However, the activity of FOPflash (black bar) remained relevantly constant in response to the treatment. This result further proved that inhibition of GSK-3β could induce the TCF/β-catenin transcriptional activity in the FLT3-ITD–expressing cells.
Expression of a Dominant Negative Form of GSK-3β Attenuated the Response of β-catenin, c-Myc, and Cyclin D3, as Well as TCF/β-catenin Driven Promoter, to the FLT3-ITD Suppression
Additional evidence that GSK-3β regulates β-catenin activity was sought using a dominant negative form of GSK-3β (dnGSK-3β with HA tag) 20,32 stably expressed in Ba/F3-FLT3-ITD cells. The FLT3 kinase activity was suppressed by increasing concentrations of PKC412 to examine the response of β-catenin, c-Myc, and cyclin D3, using 3 pairs of ITD/vector and ITD/dnGSK-3β monoclonal lines (Fig. 2C and data not shown). The expression of dnGSK-3β was evidenced by the double bands of GSK-3β blot, as well as by the HA blot (Fig. 2C, lanes 7 to 12). FLT3-ITD autophosphorylation was reduced by increasing concentrations of PKC412 in both vector- and dnGSK-3β–expressing cells, as was phosphorylation at serine 9 of GSK-3β. However, β-catenin, c-Myc, and cyclin D3 showed differential responses. In vector-expressing cells, the levels of β-catenin, c-Myc, and cyclin D3 were reduced by PKC412 in a dose-dependent pattern (Fig. 2C, lanes 1 to 6), whereas in dnGSK-3β–expressing cells, the total levels of β-catenin and cyclin D3 remained relatively constant (Fig. 2C, lanes 7 to 12). The level of c-Myc did go down, but its decline was slower than that in the vector-expressing cells.
To further demonstrate that dnGSK-3β could attenuate the response of β-catenin to the inhibition of the mutant FLT3 receptor, we analyzed the effect of PKC412 on the TCF/β-catenin–driven promoter activity using TOPflash and FOPflash reporter assay as shown in Figure 2D. The top panel shows that in dnGSK-3β–expressing Ba/F3-ITD cells, PKC412 had no inhibitory effect on TOPflash (black bar), whereas in the vector-expressing Ba/F3-ITD cells, the TOPflash activity was inhibited (gray bar). In contrast, the FOPflash reporter showed a similar response pattern to PKC412 treatment in both vector- and dnGSK-3β–expressing Ba/F3-ITD cells, as shown in the bottom panel.
Taken together, these data demonstrate that in the FLT3-ITD–expressing cells, inhibition of GSK-3β induced β-catenin and its target genes c-Myc and cyclin D3 and further rendered these proteins, along with TCF/β-catenin–driven promoter, less sensitive to the inhibition of mutant FLT3 receptor. Our results suggest that inhibition of GSK-3β is sufficient to activate β-catenin in the context of FLT3-ITD, and GSK-3β could serve as a nexus for cross-talk between FLT3-ITD and the Wnt/β-catenin pathways.
Knockdown of GSK-3β Decreases the Potencies of the FLT3 Kinase Inhibitors on Proliferation and Colony Formation in the Ba/F3-FLT3-ITD Cells, and the Potency Could Be Restored via Knockdown of Either TCF4 or β-catenin
The GSK-3β–mediated differential responses of β-catenin to PKC412 inhibition prompted us to further study the potential biological effects induced by GSK-3β inhibition in the context of FLT3-ITD mutation.
Proliferation of Ba/F3-ITD-vector and Ba/F3-ITD–dnGSK-3β cells was assayed. Although the growth rates between these 2 cell lines were similar (data not shown), their proliferation showed differential responses to the FLT3-ITD kinase inhibition (Fig. 3A). At tested concentrations, the growth of Ba/F3-ITD–dnGSK-3β cells had a reduced response to PKC412, with the most significant reduction at 15 nM (Fig. 3A, left). This observation was further confirmed by 2 additional FLT3 kinase inhibitors, AST 487 30 and FLT3 inhibitor III 31 (middle and right). Such reduced sensitivity to the FLT3-ITD inhibition was further proved by transiently expressing GSK-3β siRNA (Fig. 3B, left). The siRNA suppression of GSK-3β is shown in Figure 3C.
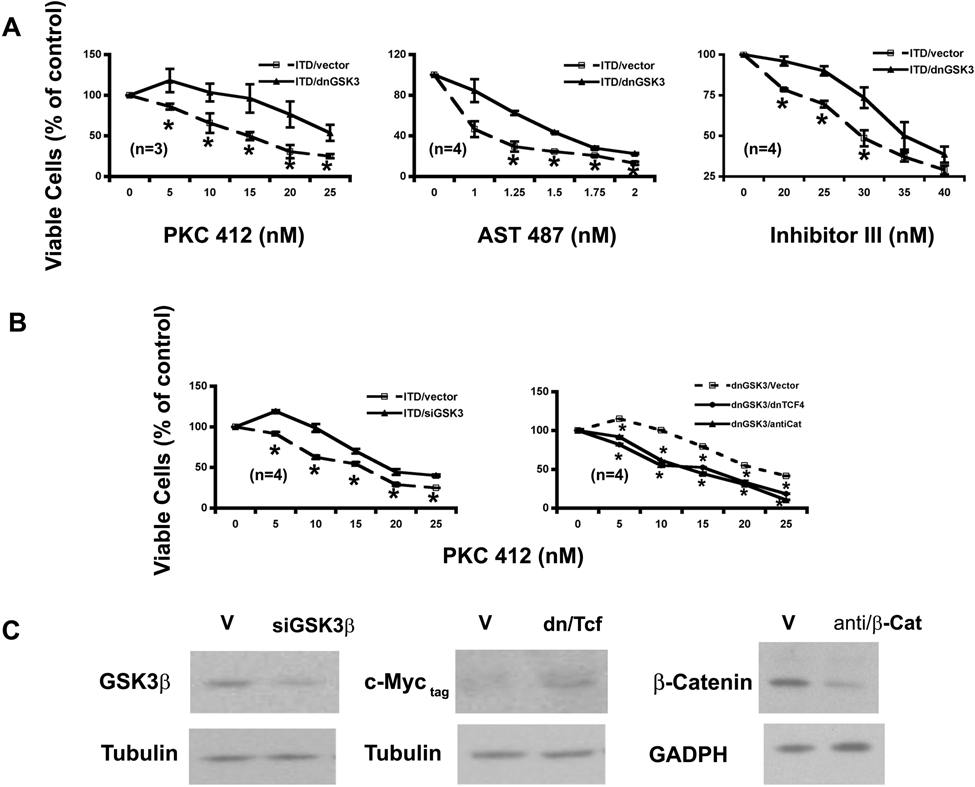
GSK-3β inhibitions attenuate the response of Ba/F3-FLT3-ITD cells to the FLT3 kinase inhibitors on cell proliferation in a Wnt/β-catenin pathway–dependent manner. (
We next examined the effect of inhibiting GSK-3β on colony formation using a soft agar assay. More colonies were formed by the cells with knockdown of GSK-3β (Fig. 4C). As in proliferation assay, the potency of PKC412 on colony formation was also reduced by inhibition of GSK-3β via either stably expressing its dominant negative form (Fig. 4A) or transiently expressing its siRNA (Fig. 4B). The most significant reduction was at 5 and 15 nM, respectively. In both assays, the IC50 of PKC412 was increased about 10 nM upon inhibition of GSK-3β. Further analysis using AST 487 and FLT3 inhibitor III confirmed that suppression of GSK-3β did render the FLT3-ITD–expressing cells less sensitive to the inhibition of FLT3-ITD on colony formation (Fig. 4A).
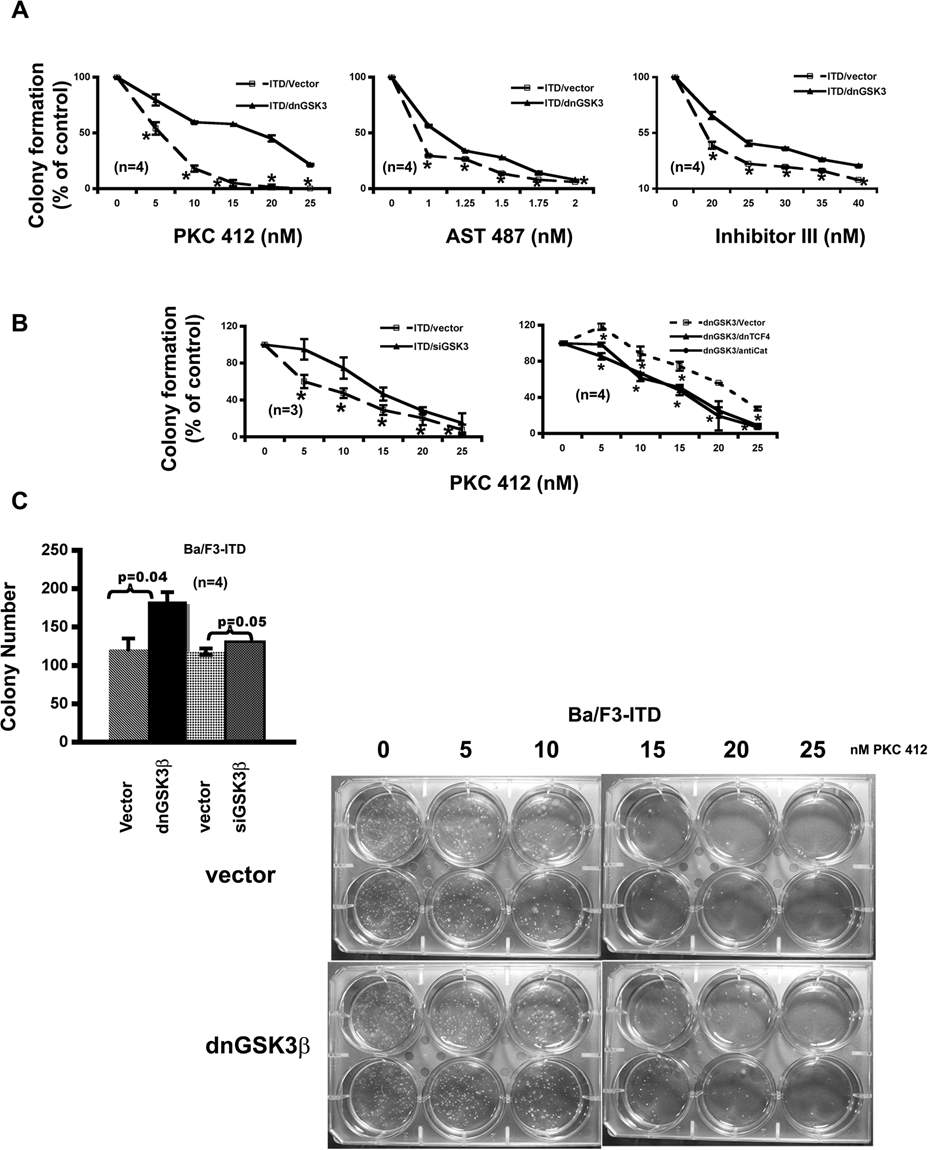
GSK-3β inhibitions attenuate the response of Ba/F3-FLT3-ITD cells to the FLT3 kinase inhibitors on colony formation in a Wnt/β-catenin pathway–dependent manner. (
To determine whether such reduced potencies of FLT3 kinase inhibitors by GSK-3β inhibition were Wnt/β-catenin pathway related, a dominant negative form of TCF4 (dnTCF4) 17 or antisense β-catenin 34 was expressed transiently in the Ba/F3-FLT3-ITD–dnGSK-3β cells. The proliferation and colony formation of the resulted cells with either expression of dnTCF4 or knockdown of β-catenin (Fig. 3C) were assayed in response to increased concentration of PKC412 (Fig. 3B and Fig. 4B, right). The results demonstrated that inhibition of either TCF4 or β-catenin brought the response curves to PKC412 back down to a similar position as that of the parental FLT3-ITD–expressing cells in both assays, suggesting that reduced potencies to the FLT3 inhibitors in the cells with inhibitions of GSK-3β could be due to the activation of the Wnt/β-catenin pathway to a large extent. Together with the biochemical evidence shown in Figures 1 and 2, these data further demonstrated that GSK-3β inhibitions activate β-catenin biologically in the context of FLT3-activating mutations.
Wnt3A Ligand Also Induces Colony Formation and Attenuates the Potencies of the FLT3 Kinase Inhibitors
The finding that endogenous activation of the Wnt/ β-catenin pathway via inhibition of GSK-3β induced colony formation and reduced the potencies of the FLT3 kinase inhibitors led us to hypothesize that exogenous activation of the Wnt pathway should have similar effects. To test this hypothesis, the Wnt3A ligand was applied in FLT3-ITD–expressing cells and then their proliferation and colony formation were assayed in response to 3 FLT3 inhibitors. Wnt3A by itself had little direct effect on proliferation of the FLT3-ITD–expressing cells (data not shown) but did increase the colony formation (Fig. 5C). As expected, Wnt3A stimulation reduced the potencies of these inhibitors on both proliferation (Fig. 6A, B, and C) and colony formation (Fig. 5A and B) in the cells either ectopically expressing FLT3-ITD or derived from AML patient samples harboring FLT3-ITD mutation.
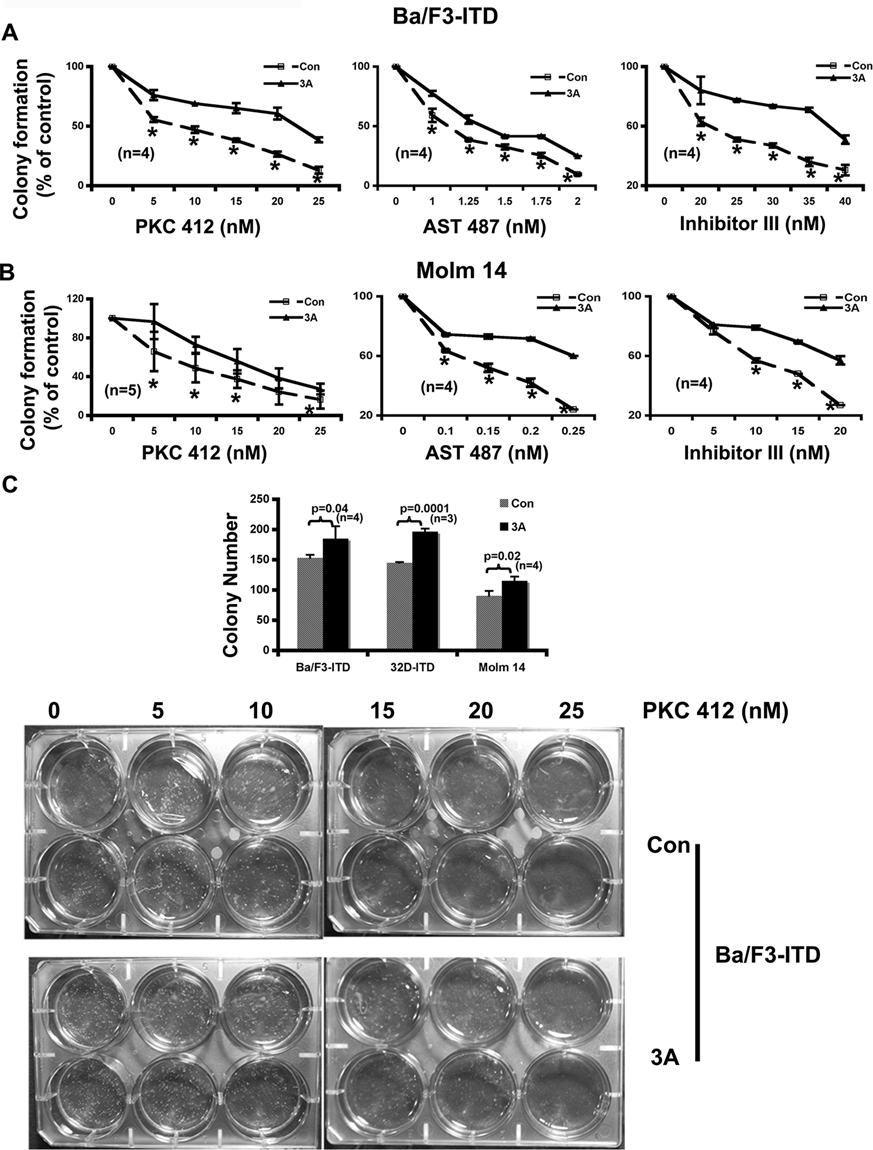
Exogenous activation of the Wnt/β-catenin pathway by the Wnt3A ligand attenuates the potencies of the FLT3 kinase inhibitors on colony formation. (
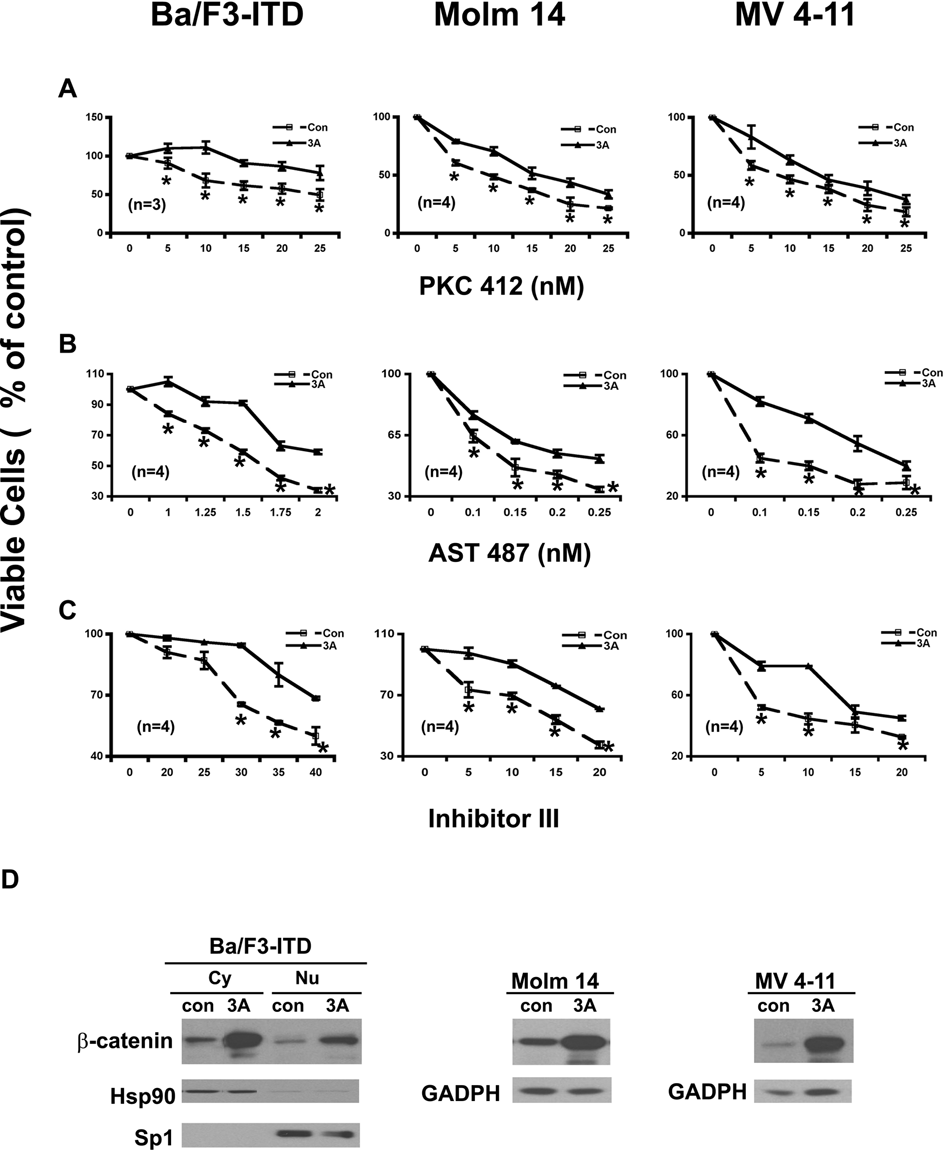
Exogenous activation of the Wnt/β-catenin pathway by the Wnt3A ligand attenuates the potencies of the FLT3 kinase inhibitors on cell proliferation in either Ba/F3 cells ectopically expressing FLT3-ITD or cell lines derived from acute myeloid leukemia (AML) patient samples harboring FLT3-ITD mutations. (
The analysis of β-catenin level on the stimulation of the Wnt3A ligand (Fig. 6D) demonstrated that the cells did respond to the Wnt3A stimulation and that the observed biological effects of the Wnt3A ligand stimulation were due to the activation of the Wnt/β-catenin pathway.
Taken together, these results demonstrated that in the FLT3-ITD–expressing cells, activation of the Wnt/β-catenin pathway, both endogenously and exogenously, reduced the potencies of the FLT3 kinase inhibitors and further suggested a critical role of GSK-3β in the cross-talk between these 2 pathways.
Discussion
In this report, we demonstrate the importance of GSK-3β in mediating the induction of β-catenin in response to FLT3-ITD mutations, which culminates in the reduction of the potencies of the FLT3 kinase inhibitors. Activation of the Wnt/β-catenin pathway has been shown to increase self-renewal of both normal hematopoietic stem cells 8 and the progenitor cells from patients with chronic myelogenous leukemia 35 and other tumors. 36-40 Our findings point to an importance of GSK-3β in the pathogenesis of AML.
Although GSK-3β is an important component in the β-catenin destruction complex, 13,14 it is reported that both GSK-3β kinase activity and its access to β-catenin 21,41 are equally important and that GSK-3β regulation in β-catenin is cell type specific. 20,21 Using known targets of the Wnt/ β-catenin pathway, as well as TCF/β-catenin responsible promoter, we demonstrated that inhibitions of GSK-3β resulted in the activation of the Wnt/β-catenin pathway in a mutant FLT3 receptor and PI3 kinase pathway–dependent manner, both biological and biochemical, which suggests that in our system, inhibition of GSK-3β alone is sufficient to activate β-catenin.
An interesting observation in our study is that the potencies of the FLT3 inhibitors were decreased upon activation of the Wnt pathway by either endogenous inhibition of GSK-3β or exogenous stimulation with the Wnt3A ligand. This finding suggests that the cross-talk between the FLT3-ITD and the Wnt pathways may induce partial drug resistance to the inhibitors of this kinase and increase the tolerances of the host cells to cytotoxic agents. A recent study has suggested that interaction of the Wnt pathway with the adhesion-dependent pathway could control the chemosensitivity of AML mediated by GSK-3β and NF-κB. 42 Although the mechanism of such increased resistance remained unclear, our findings suggest that the Wnt/β-catenin activation in the context of the FLT3-activating mutations may culminate in genome instability. It would be interesting to find out whether the FLT3-activating mutations could induce a secondary mutation via activation of β-catenin.
Materials and Methods
Reagents, Cell Lines, and Antibodies
GSK-3β inhibitor VII, FLT3 inhibitor III, Ly 294002, Wortmannin, and protease inhibitor cocktail set I were from Calbiochem (San Diego, CA). PKC412 and AST 487 were kindly provided by Dr Johannes Rossel (Novartis Pharma, Switzerland). Lithium chloride was from Sigma (St. Louis, MO). NE-PER nuclear and cytoplasmic extraction reagents were from PIERCE (Rockford, IL). SELECT Agar was from Invitrogen/Gibco (Carlsbad, CA). L cells, L/Wnt3A cells, and U937 cells were from ATCC (Manassas, VA). The following antibodies were used: pTyr(pY99), FLT3(c-20), c-Myc(N-262), c-Myc(9E10), β-catenin(E-5), GSK-3β(H 76), cyclin D3(c-16), TCF4(H125), and Sp1(PEP 2), from Santa Cruz (Santa Cruz, CA); pAKT(Ser 473), AKT, pGSK-3β(ser 9), GADPH, and β-catenin, from Cell Signaling (Danvers, MA); Hsp 90(clone AC 88) from Stressgen (Victoria, BC, Canada); and Tubulin(clone DM 1A) from Sigma.
Cell Culture
Ba/F3-FLT3-ITD, Ba/F3-FLT3-N841I, and 32D-FLT3-ITD cells were cultured in the regular RPMI 1640 medium containing 800 μg/mL G418. 5 Ba/F3-pClneo cells were cultured in the same medium but supplemented with 10% of the WEHI3B conditioned medium (as a source of IL3). The Molm 14 and MV4-11 cells were cultured in the regular RPMI 1640 medium. The culture media for U937 cells, L cells, and L/Wnt3A cells were as per the manufacturer’s protocols (ATCC). The cells were maintained in a humidified incubator at 37°C with 5% CO2.
Western Blotting Analysis
Total proteins were isolated using NP-40 buffer following the standard protocol. 43 For separation of cytoplasmic and nuclear protein, the cells were lysed in the NE-PER nuclear and cytoplasmic extraction reagents per the manufacturer’s protocol. Protein extracts were then subjected to Western blotting analysis using the antibodies indicated.
Colony Formation Assay
SELECT agar was used for colony formation assay. 44 Briefly, cells resuspended in culture medium with 0.4% agar and drug treatments (top agar) were seeded duplicate in a 6-well plate containing 0.5% agar in the culture medium with the same drug treatment as those for the top agar. Colonies were counted on days 10 to 14.
Generation of Stable Cell Lines
Dominant negative GSK-3β (K85A mutant) 32 was subcloned into pBABE/puro expression vector 45 to facilitate selection. FLT3-ITD 46 was subcloned into pCIneo (Promega, Madison, WI) expression vector. pCIneo/FLT3-N841I was generated by site-directed mutagenesis. 5 Plasmid DNA was introduced into host cells by electroporation using Gene Pulser Xcell (Bio-Rad, Hercules, CA). Twenty-four to 36 h after electroporation, the cells were grown in the selective medium with appropriate antibiotics (800 μg/mL for neomycin, 2 μg/mL for puromycin) for selection.
Transient Expression Assays
siRNA of GSK-3β was purchased from SABiosciences (Frederick, MD) and manipulated following the manufacturer’s protocol. The cDNAs of dominant negative TCF-4 17 and antisense–β-catenin 34 were used directly without further subcloning. Both Gene Pulser Xcell and the Nucleofector Device (Amaxa Inc., Gaithersburg, MD) were used for introducing target constructs into host cells with 50% to 80% transfection efficiency. The cells were analyzed 20 to 24 h after electroporation.
TOPflash reporter and FOPflash reporter (Upstate/Millipore, Lake Placid, NY) were introduced into host cells by electroporation using Gene Pulser Xcell. Twenty to 24 h after electroporation, the cells were treated and analyzed for luciferase activity using the Dual-Luciferase Reporter Assay Kit (Promega).
Footnotes
Acknowledgements
The authors thank Dr Jim Woodgett (University of Toronto, Canada), Dr Bert Vogelstein and Dr Kenneth W. Kinzler (The Johns Hopkins Medical Institutions & Howard Hughes Medical Institute, Baltimore, MD), Dr Jane Trepel (National Institutes of Health/National Cancer Institute, Bethesda, MD), and Dr Ellen Weisberg, Dr Blanca Scheijen, Dr Yuki Yuza, and Dr Matthew Meyerson (Dana-Farber Cancer Institute, Boston, MA) for providing reagents and constructs. They also thank Dr Heidi Greulich and Dr Sarah Walker (Dana-Farber Cancer Institute) for the protocols and suggestions on the colony formation and luciferase assays.
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the National Institutes of Health (CA66996, CA36167, and DK50654; J.D.G.) and by a Specialized Center of Research Award from the Leukemia and Lymphoma Society (J.D.G.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.