
Abstract
Adenosine monophosphate–activated protein kinase (AMPK) is a metabolic regulator that promotes energy conservation and restoration when cells are exposed to nutrient stress. Given the high metabolic requirement of cancer cells, AMPK activation has been suggested as a potential preventative and therapeutic target. However, previous findings have shown that AMPK activity is diminished in some cancers. Expression of the 2 catalytic isoforms, AMPKα1 and AMPKα2, was evaluated in primary breast cancer and matched nontumor-adjacent tissue samples using immunohistochemistry. AMPK-dependent growth signaling events were examined in primary human mammary epithelial cells (HMECs) using RNAi to understand the importance of AMPKα2 in normal growth regulation. To test whether AMPKα2 would reinstate growth control and apoptotic mechanisms in breast cancer cells, metabolic stress assays and tumor xenografts were performed in MCF-7 cells, expressing low levels of AMPKα2, with stable transfection of either green fluorescent protein (GFP) or AMPKα2 expression constructs. AMPKα2 was found to be significantly suppressed in breast cancer tissue samples, whereas AMPKα1 was not. In normal HMECs, low glucose stress resulted in AMPK-driven growth inhibition. Interestingly, this response was ablated when AMPKα2 was silenced. Metabolic stress assays in MCF-7 cells indicated that AMPKα2 expression reduced both mTOR signaling and cyclin D1 expression, contributing to G1-phase cell cycle arrest. Cells expressing AMPKα2 underwent apoptosis more readily than GFP control cells. Xenograft studies demonstrated that MCF-7 tumors expressing AMPKα2 display reduced proliferation and increased apoptotic events. Furthermore, AMPKα2 xenografts exhibited diminished cyclin D1 levels along with an increased amount of nuclear p53, thereby implicating the AMPKα2-p53 signaling axis as a mediator of cell apoptosis. Together, these results highlight the significance of reduced AMPK activity contributing to human carcinogenesis and, specifically, the role of AMPKα2 with respect to its control of normal mammary epithelial cell growth and its reduced expression in breast cancer.
Introduction
Adenosine monophosphate–activated protein kinase (AMPK) is a metabolic sensor that has emerged as a critical target in cancer biology. AMPK functions as a heterotrimeric protein comprising α, β, and γ subunits. 1 When a cell is exposed to nutrient deprivation or hypoxic stress, the catalytic AMPK α subunit is phosphorylated at threonine 172 by upstream kinases (LKB1, CAMKKβ, and TAK1) and allosterically activated through AMP binding to the regulatory γ subunit.2,3 Subsequent AMPK signaling events restore metabolic balance by decreasing energy consumption through reduced protein, lipid, and fatty acid synthesis and instead by increasing energy production by upregulating glucose and fatty acid uptake, glycolysis, fatty acid oxidation, and promotion of angiogenic regulators such as VEGF.4-6 AMPK also mediates cell fate by inducing apoptosis through the direct phosphorylation of p53 at serine 157-9 or by promoting cell survival through ULK1/autophagy activation.10,11 Together, AMPK signaling events protect cell integrity by preventing cell cycle progression when insufficient resources are available.
Previous studies indicate that AMPK may prevent tumorigenesis through a variety of mechanisms. 12 Its upstream kinase, LKB1, is a proven tumor suppressor that coordinates cell polarity in part through AMPK signaling. Disruptions in cell polarity lead to disorganized cell division due to cross-talk with the pro-proliferative WNT pathway and growth-restrictive AMPK-mTOR metabolic pathway. As a result, individuals with LKB1 germline mutations are predisposed to a variety of carcinomas including gastrointestinal, pancreatic, ovarian, and breast. 13 Similarly, AMPK has emerged as a critical regulator of chromosome fidelity. Immunofluorescence studies in multiple cell types showed that activated AMPK localizes to the centrosomes throughout mitosis and transfers to the cleavage furrow during cytokinesis. 14 When the AMPKα ortholog (SNF1A) is knocked out in Drosophila melanogaster, it was seen that the loss of AMPK causes mitotic defects within the embryos, including lagging chromosomes and aneuploidy, which could ultimately result in tumorigenesis. 15 Furthermore, a recent study performed in preneoplastic JB6 Cl41 mouse epidermal cells demonstrated the importance of AMPK in preventing tumor initiation in which both AMPK overexpression and AICAR-mediated AMPK activation inhibited EGF-driven transformation, while downregulation of AMPK activity through a Pin1-PP2A complex led to the induction of transformed cells. 16 Together, these characteristics provide evidence for AMPK’s role in cellular metabolic sensing and tumor suppressor–like activity in normal tissues.
In addition to its proposed role in reducing tumor initiation, AMPK activation has also been investigated as a potential cancer therapy through the application of the antidiabetic drug metformin (N,N-dimethylbiguanide). It is proposed that metformin inhibits tumor growth by reducing serum glucose levels and insulin/insulin-like growth factors as well as through intratumoral AMPK activation.17,18 In fact, a 2005 retrospective study determined that the cancer incidence among type II diabetic patients receiving metformin therapy was significantly reduced compared to patients receiving other treatment regimens. 19 Experimental cancer models confirmed these notions and indicated that metformin dosing was critical to its neoplasia-reducing effects.20-24 These findings suggest that chronic administration of metformin may reduce cancer formation and also implicate AMPK activation as a possible strategy for the treatment of primary cancers.25,26
Interestingly, a recent report showed that AMPK activity was absent in 90% of the primary breast cancer cases that were examined. 27 This study analyzed 354 specimens, with immunohistochemistry revealing low levels of phosphorylated AMPKαThr172 as well as the AMPK substrate acetyl co-A carboxylase (p-ACC), in tumors compared to the normal breast epithelium. Given this finding, it is crucial to further examine AMPK expression in primary breast cancer to determine whether the AMPK activation potential may be limited in early breast cancer or breast cancer subtypes.
Here, we sought to evaluate the expression of both catalytic subunits, AMPKα1 and AMPKα2, in primary breast cancer samples. Our findings revealed that AMPKα2 was widely suppressed in breast cancer, leaving the AMPKα1 subunit as the major contributor towards AMPK signaling. Silencing AMPKα2 in primary HMECs hindered their ability to halt proliferation signals under nutrient stress, suggesting that AMPKα2 may selectively provide a tumor suppressor–like role. Accordingly, it was found that exogenous expression of AMPKα2 in the estrogen-dependent breast cancer MCF-7 cell line restored metabolic growth control through cell cycle checkpoints and induction of apoptosis.
Results
AMPKα2 is suppressed in primary human breast cancer
Primary human breast cancers were evaluated for AMPK α isoform expression in situ to better understand their potential signaling contribution to total AMPK activity. A cohort of 40 tumor samples and their corresponding patient-matched, nontumor-adjacent (ADJ) breast epithelial tissue and an additional 35 samples of unrelated normal tissue sections were acquired from patients undergoing surgical resection or voluntary reduction mammoplasty. Immunohistochemistry staining of AMPKα1 and AMPKα2 was performed on serial tissue sections, and the staining was scored by 3 independent reviewers. The normal mammary tissue cohort revealed strong, uniform expression of both AMPKα1 and AMPKα2 within the ductal epithelium, thus establishing the presence of both isoforms within normal breast tissue (Fig. 1A). Analysis of these isoforms in the patient-matched ADJ and tumor tissue cohort found that the AMPKα1 protein was abundantly expressed in both tissue types; however, AMPKα2 protein expression was significantly reduced in all tumors compared to their patient-matched ADJ samples (Fig. 1B). AMPKα2 protein expression was reduced by 27% in tumor samples compared to the patient-matched ADJ samples and by 37% when compared to the normal epithelial tissue samples (Fig. 1C). To further demonstrate this point, 2 independent tissue core arrays were evaluated with combined 140 neoplastic breast samples provided in duplicate (Pantomics Inc., San Francisco, CA). Within these samples, AMPKα2 expression was also found to be significantly suppressed in all cancer grades compared to normal, hyperplastic, or fibroadenoma tissues (Suppl. Fig. S1A). There was no selectivity for tumor grade, lymph node status, or patient age (Suppl. Fig. S1A-C). Together, these data reveal a significant and widespread reduction of AMPKα2 expression in mammary epithelial carcinoma.
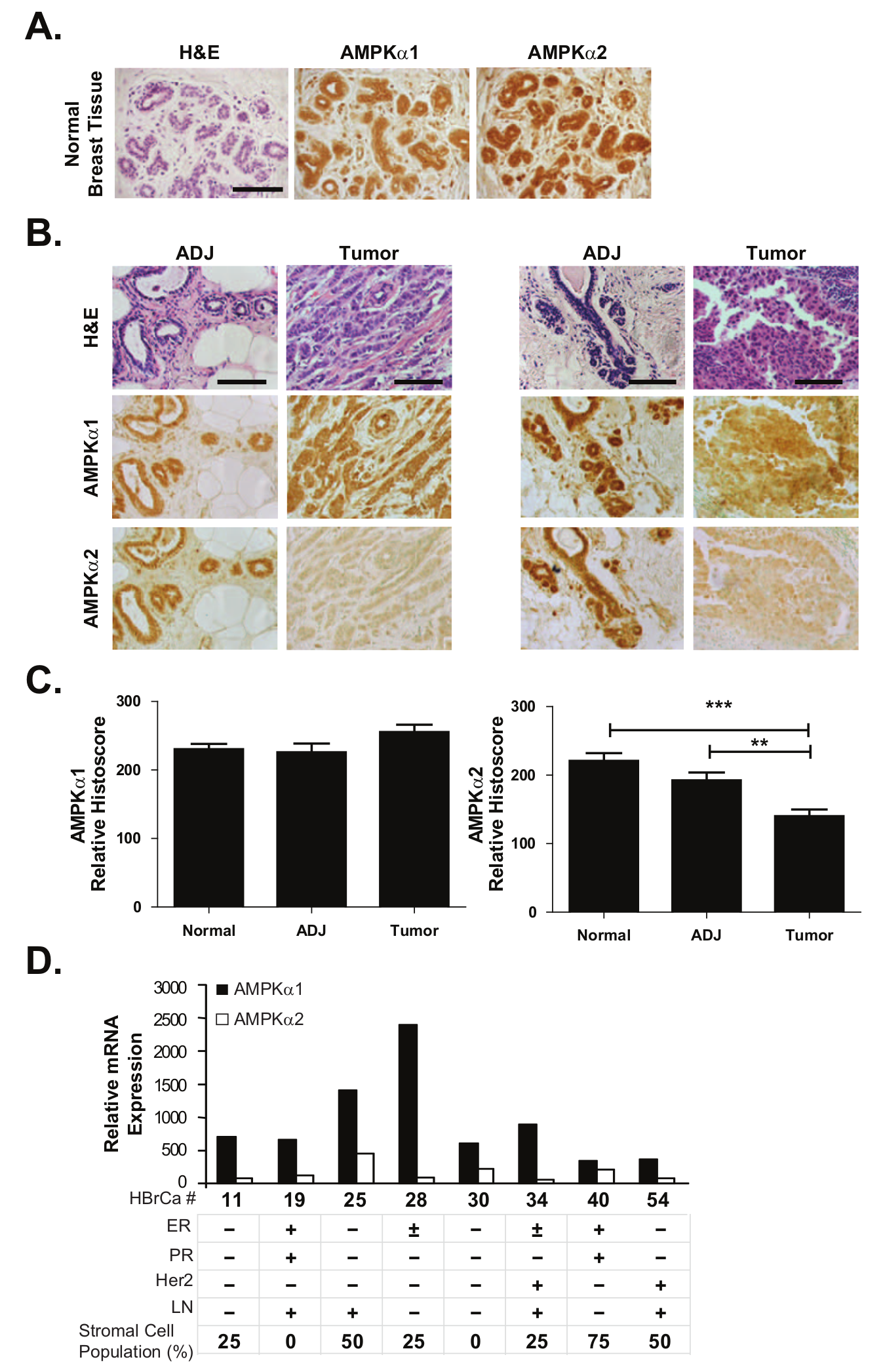
AMPK α isoform expression in noncancerous and cancerous primary human breast tissues. Immunohistochemistry was used to evaluate AMPK α isoform expression in a cohort of 40 breast tumor samples and their patient-matched ADJ epithelial tissues. An additional 35 normal breast epithelial tissue samples from unrelated patients were also assessed. (
A comprehensive Illumina array profile of 8 primary human breast cancers was then used to evaluate whether the observed AMPKα2 suppression was occurring at the mRNA level. RNA was extracted from tissue cores taken from dense tumor regions as guided by H&E-stained frozen sections. Notably, AMPKα2 mRNA transcripts were uniformly lower than AMPKα1 in each patient case (Fig. 1D). Of the samples retaining the highest AMPKα2:AMPKα1 mRNA expression ratios (HBrCa-25, HBrCa-30, and HBrCa-40), 2 of them had significant stromal cell contamination as estimated from H&E staining: HBrCa-25 = 50% and HBrCa-40 = 75% of nontumor stromal cellularity. The remaining samples contained only 0% to 25% of stromal cell contamination (with the exception of HBrCa- 54 at 50%), implying that the AMPKα2:AMPKα1 mRNA expression ratio decreases in the epithelial tumor cell compartment, thereby confirming the immunohistochemistry observations. Overall, these data suggested that the reduced AMPKα2 staining observed in the patient-matched tissue cohort was primarily due to the modulation of expression at the RNA level.
Primary HMECs exhibit AMPK-dependent growth regulation
To understand the importance of AMPK signaling in normal mammary epithelial growth regulation, primary human mammary epithelial cells (HMECs) were employed. Two independent HMEC preparations (HMEC-1 and HMEC-2) were found to express both AMPKα1 and AMPKα2 proteins, as determined by immunoblots, consistent with our immunohistochemistry findings in the nontumorigenic mammary epithelium (Fig. 2A). When cultured under low glucose conditions, both cell populations demonstrated strong AMPK activation at the AMPKαThr172 phosphorylation site (Fig. 2B). In addition to AMPK activation, HMECs cultured under low glucose displayed reduced p-p70S6K and cyclin D1 protein levels, indicating that the AMPK signaling pathway exerts a metabolic growth control over protein synthesis and cell proliferation pathways. The effect of nutrient restriction signaling on growth control was further supported in functional growth assays, showing that low glucose significantly inhibited cell proliferation in both HMEC-1 and HMEC-2 over 3 days compared to control media (Fig. 2C).
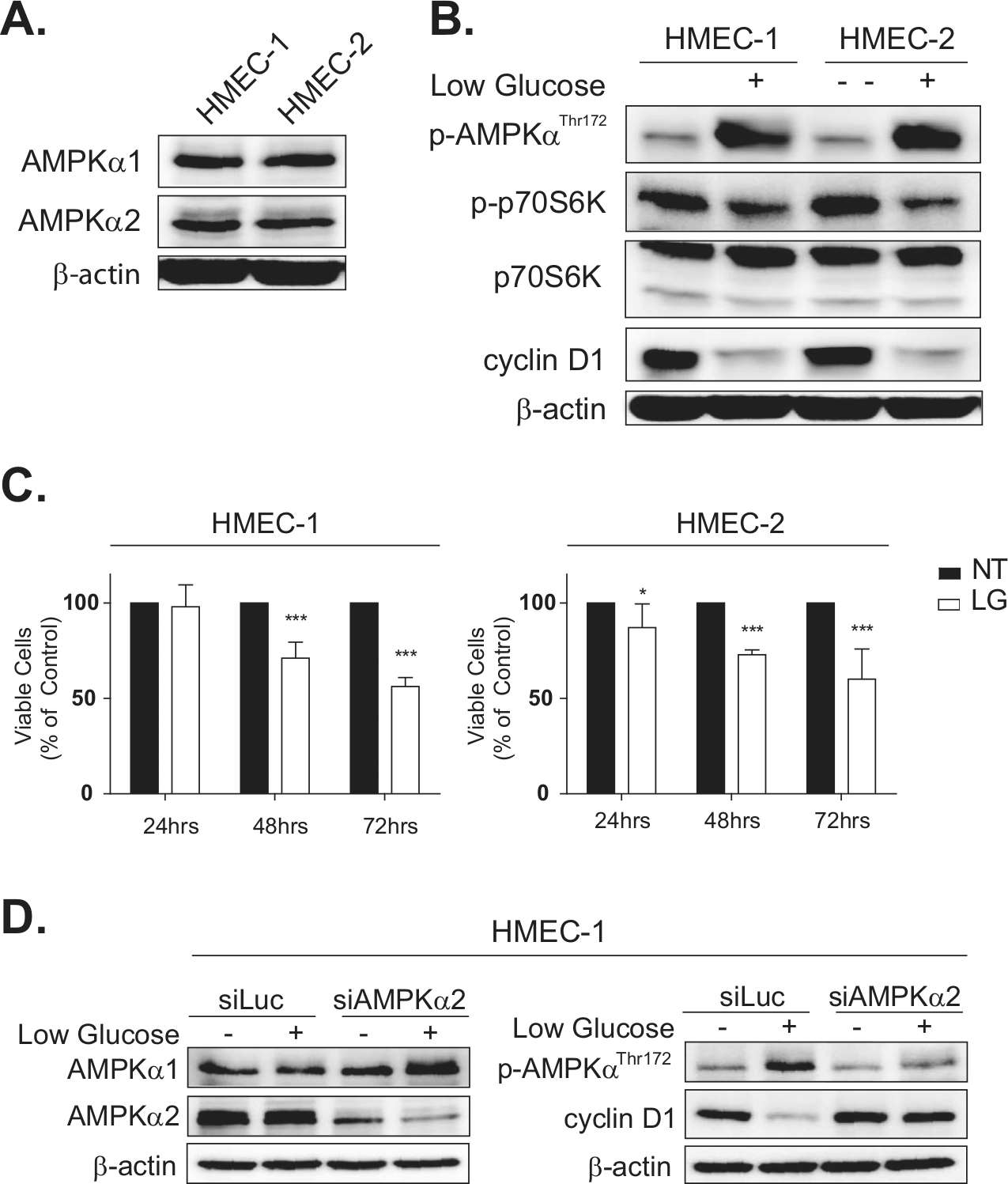
AMPK-mediated growth regulation in primary mammary epithelial cells. Two independent primary HMECs (HMEC-1 and HMEC-2) were cultured for 18 hours under no treatment (NT) or low glucose (LG) stress conditions and evaluated for AMPK signaling responses. (
To confirm that the observed growth inhibition was specific to AMPKα2 signaling mechanisms, RNAi technology was used to silence AMPKα2 expression in primary HMECs. It was expected that the loss of AMPKα2 would attenuate the growth regulation previously observed under low glucose conditions. HMEC-1 and HMEC-2 cells were transiently transfected with either siLuciferase, as a nontarget control, or pooled siAMPKα2 oligomers. HMECs transfected with constructs targeting AMPKα2 displayed an 81% reduction of AMPKα2 protein levels by 72 hours as determined by immunoblot densitometry normalized to β-actin (Fig. 2D). AMPKα1 expression was not affected by siAMPKα2, nor did siLuciferase alter AMPKα1 or AMPKα2 levels in the control cells. As predicted, AMPKα2 silencing resulted in reduced AMPK activation under low glucose; siAMPKα2 HMECs yielded less than half the amount of p-AMPKThr172 compared to siLuciferase HMECs (Fig. 2D). This was accompanied by a significant lack of reduction of cyclin D1 protein levels in the siAMPKα2-treated HMECs under low glucose conditions (Fig. 2D). Together, these data indicate that AMPKα2 plays a critical role in regulating mammary cell growth signals, and suppression of total active AMPK affects downstream regulators including cyclin D1 as described previously.28,29
MCF-7 cancer cell line expresses low levels of AMPKα2 protein
A panel of established human breast cancer cell lines was selected to evaluate the presence of AMPKα1 and AMPKα2 expression in experimental model systems. Immunoblot analysis of both AMPK catalytic isoforms found that AMPKα1 was consistently expressed in all cell lines, whereas AMPKα2 exhibited differential expression levels depending on the cell type examined (Fig. 3A). Both MCF-7 and MDA-MB-231 cells displayed low levels of AMPKα2 protein expression, while T47-D, BT-474, and MDA-MB-468 cells displayed more abundant levels.
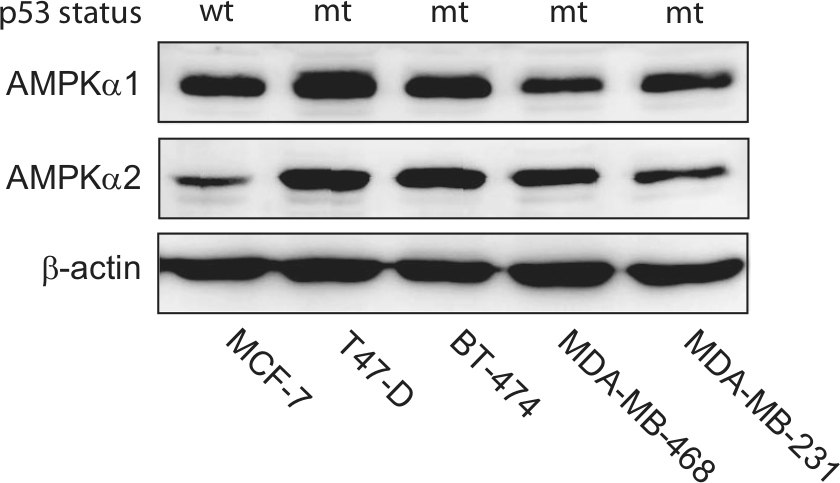
AMPK α isoform expression in breast cancer cell lines. Human breast cancer cell lines (MCF-7, T47-D, BT-474, MDA-MB-468, and MDA-MB-231) were cultured under basal growth conditions and evaluated for AMPK α isoform expression. (
AMPKα2 re-expression in MCF-7 cells
The observed loss of AMPKα2 expression in primary breast cancers indicates that its presence in the normal mammary epithelium may inhibit tumor initiation and/or development. To study whether AMPKα2 expression could affect the tumorigenic properties in transformed breast cancer cells, MCF-7 cells were used to re-express the AMPKα2 isoform. This cell line was chosen due to the minimal amount of endogenous AMPKα2 protein expression (Fig. 3A) as well as its wild-type p53 status. 30 MCF-7 cells were stably transfected with the AMPKα2 isoform containing a C-terminal V5 epitope tag (AMPKα2-V5) or with a green fluorescent protein (GFP) control vector. Immunoblot analysis confirmed the expression of exogenous GFP and AMPKα2 proteins (Fig. 4A). Control cells displayed strong GFP expression while maintaining high AMPKα1 and null AMPKα2 expression, indicating that transfection and selection did not alter the AMPK α isoform ratio in the control cells. In comparison, cells expressing AMPKα2 exhibited a 2-fold increase in the total AMPK α isoform expression as determined by densitometry from respective immunoblots (data not shown). Interestingly, the expression of AMPKα2 appeared to negatively regulate the expression of the AMPKα1 isoform when compared to the GFP control cells. The functionality of the exogenous AMPKα2 protein was also verified by immunoprecipitation using an anti-V5 antibody to isolate AMPKα2-V5 and subsequently detected using an anti–p-AMPKαThr172 antibody to ensure its ability to be phosphorylated under hypoxic stress (data not shown). To determine whether the established cell lines displayed phenotypic growth differences, cells were cultured under basal conditions, and growth was monitored over 72 hours. It was observed that AMPKα2 cells had a slight decrease (11.3%, P ≤ 0.05) in growth at 72 hours compared to GFP control cells, suggesting that AMPKα2 signaling may exert minimal but significant downstream antiproliferative effects (Fig. 4B).
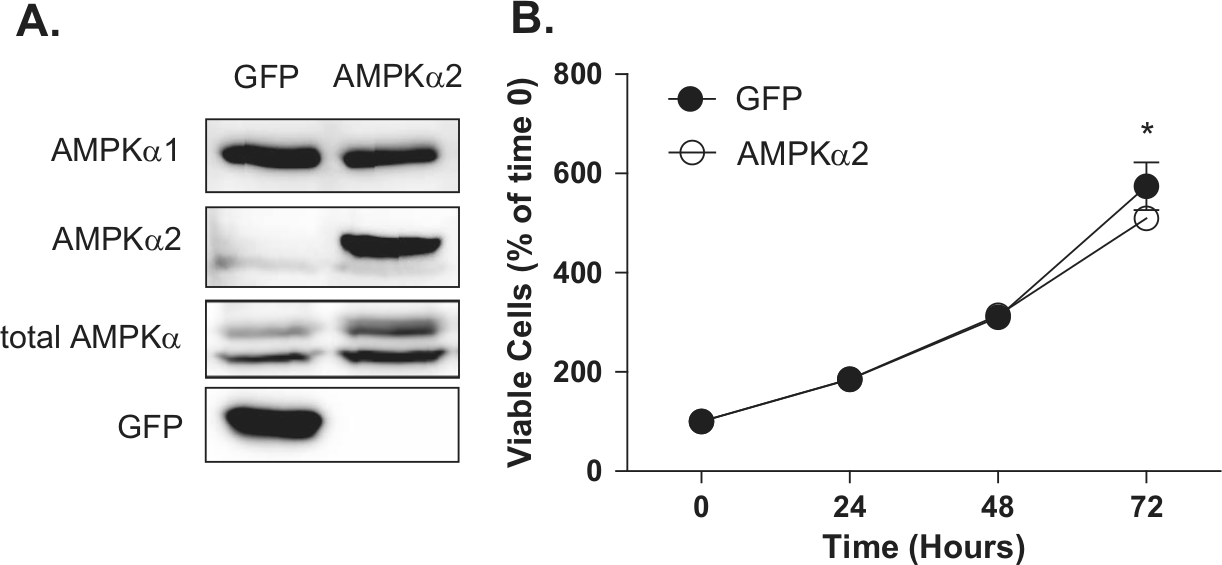
AMPKα2 re-expression in MCF-7 cells. (
AMPKα2 expression induces cell cycle arrest and apoptosis in MCF-7 cells
To evaluate whether AMPKα2 re-expression may hinder protumoral signaling events in an established cancer cell line, MCF-7 cells were used to evaluate AMPK signaling under basal or low glucose stress culture conditions in vitro. While AMPK activation was observed at similar levels in both GFP and AMPKα2 cells after 48 hours of low glucose (Fig. 5A), cells expressing the AMPKα2 isoform demonstrated stronger downstream suppression of the mTOR axis as shown by the nearly complete loss of phosphorylated p70S6K (Fig. 5B). Low glucose conditions resulted in the repression of cyclin D1 protein level and induction of p53 protein expression and cleavage of PARP in both cell types (Fig. 5C); however, this was accentuated in AMPKα2 cells. Additionally, expression of the p53 protein was increased in AMPKα2 cells under control conditions as well. Consistent with this evidence of reduced protein synthesis and proliferation, cell cycle analysis by flow cytometry revealed a 23.0% increase in G1-phase cell cycle arrest in which 55.9% ± 0.4% of AMPKα2 cells accumulated in G1 compared to only 45.4% ± 0.6% of GFP control cells (Fig. 5D). Together, these results suggest that AMPKα2 reinstates a nutrient stress growth control mechanism that was previously lost in MCF-7 cancer cells and effectively results in cell cycle arrest and apoptosis.
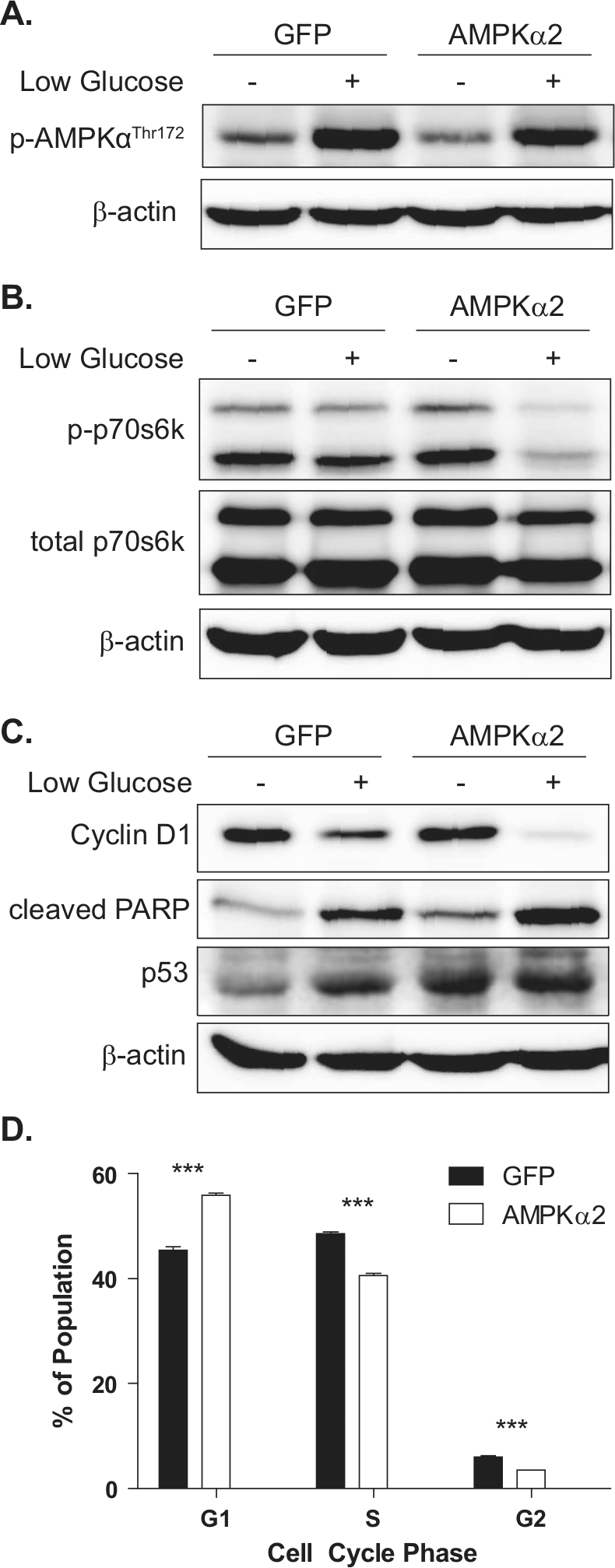
In vitro analysis of MCF-7 cells expressing AMPKα2. Immunoblot analysis was used to evaluate downstream signaling events after GFP and AMPKα2 cells were cultured under basal or low glucose conditions for 48 hours. (
AMPKα2 re-expression in MCF-7 cells inhibits tumor growth in vivo
The tumorigenicity of MCF-7 cells with minimal or high levels of AMPKα2 expression was then evaluated in vivo. Orthotopic xenografts were established in the mammary fat pad of athymic nude mice and allowed to expand for 36 days. Markedly, the tumor burden of AMPKα2 xenografts was significantly reduced compared to the control GFP-expressing group. At harvest, AMPKα2 tumors were 56.4% (P ≤ 0.001) smaller by volume and 40.3% (P ≤ 0.05) smaller by weight (Fig. 6A and 6B). Both tumor groups contained dense, viable cells as evident by H&E staining, thereby eliminating gross necrosis as a contributing factor in the observed size differences (Fig. 6C). Additionally, RNA extracted from fresh-frozen tissue cores was analyzed to confirm that the stable expression of AMPKα2 was maintained over the course of tumor expansion (Fig. 6D). Consistent with our in vitro findings, the AMPKα2-expressing tumor cells continued to show moderate downregulation of AMPKα1 RNA, thereby indicating a potential negative feedback regulation mechanism between the 2 AMPK α isoforms.
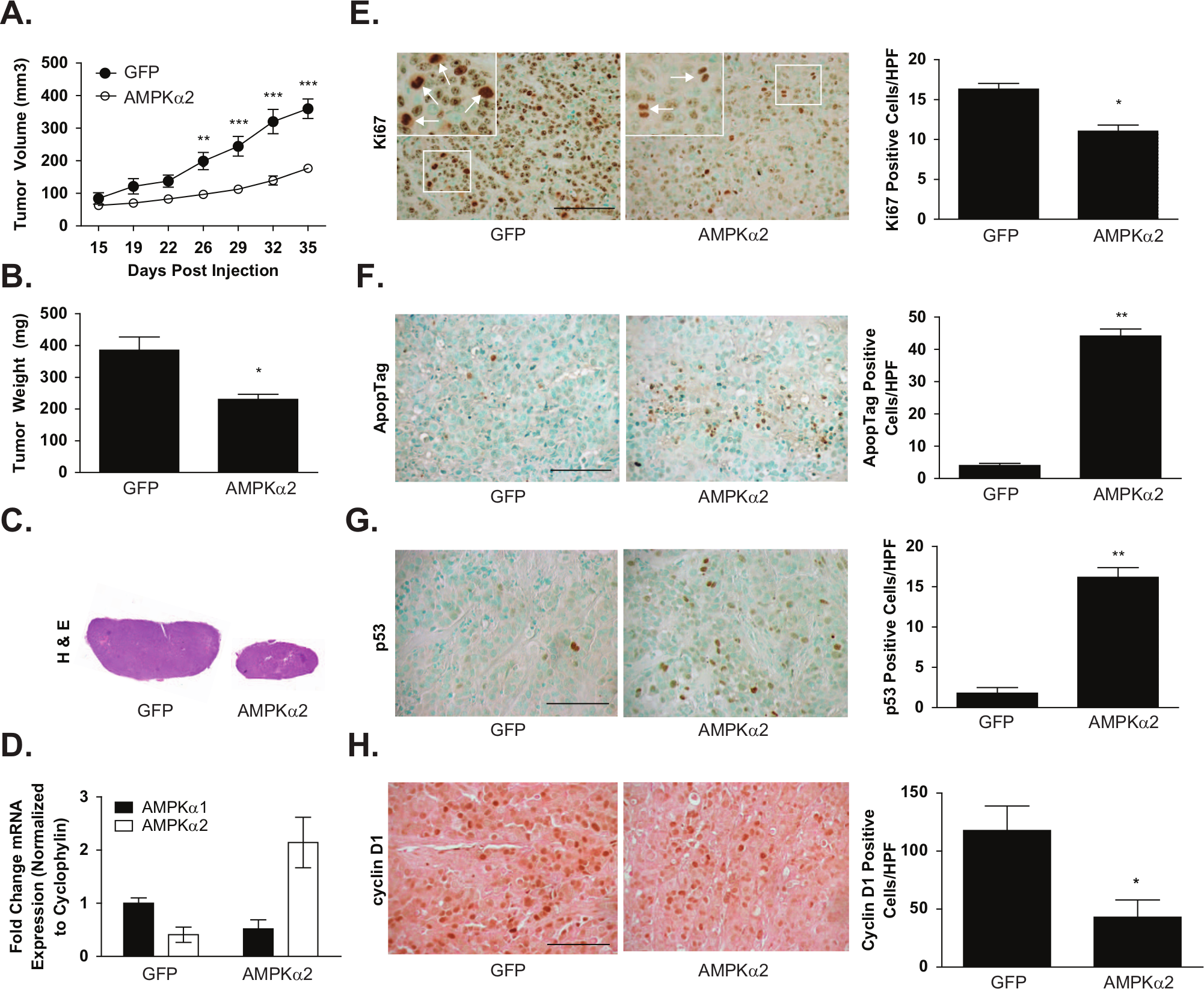
Analysis of MCF-7 xenografts with GFP or AMPKα2 expression. 8.33 × 105 MCF-7 cells with stable GFP or AMPKα2 expression were injected subcutaneously into the mammary fat pad of athymic nude mice (n = 9 and 10 animals, respectively). (
To confirm that GFP and AMPKα2 cells do not differentially respond to the effects of estradiol, growth assays were completed with media supplemented with charcoal-stripped serum (CSS) and CSS with estradiol added (Suppl. Fig. S2). The growth of the AMPKα2 cells was consistently lower when compared to GFP cells in all conditions. Specifically, treatment with CSS similarly reduced the cell growth of both GFP and AMPKα2 cells. Supplementation of CSS media with estradiol restored the growth of GFP and AMPKα2 cells to control levels in both high glucose and low glucose basal media, suggesting that GFP- and AMPKα2-overexpressing cells respond to estradiol equally.
To further investigate the potential mechanisms responsible for suppressing tumor growth in the AMPKα2 tumors, immunohistochemistry was used to evaluate proliferation and apoptotic events by Ki67 and ApopTag staining, respectively (Fig. 6E and 6F). Ki67 staining identified a 32.4% (P ≤ 0.05) reduction of proliferating cells in the AMPKα2 tumors compared to the GFP control group. Conversely, AMPKα2 expression correlated with an 11-fold (P ≤ 0.01) increase in tumor cell apoptosis, indicating that both proliferation and apoptotic rates were affected. To investigate whether the apoptotic induction could stem from an AMPK-p53 signaling pathway, as suggested by in vitro data and previous studies,7,9 p53 stabilization was probed by immunohistochemistry on paraffin tissue sections. Interestingly, tumors with AMPKα2 expression revealed a 9-fold (P ≤ 0.01) increase in positive nuclear p53 cells compared to control tumors (Fig. 6G). Levels of cyclin D1 were also assessed, and consistent with in vitro data, a significant decrease of cyclin D1 levels by 63.5% (P ≤ 0.05) in AMPKα2 tumor samples was found when compared to GFP control tumor samples (Fig. 6H). Angiogenesis did not appear to play a significant role since there were no significant differences in the intratumoral blood vessel density as determined by PECAM-1/CD31 staining (GFP: 17.6 ± 3.1 vessels/HPF; AMPKα2: 13.6 ± 1.6 vessels/HPF) or VEGF mRNA expression as determined by quantitative RT-PCR (GFP: 1.3 ± 0.2; AMPKα2: 1.1 ± 0.4).
Discussion
AMPK activation has gained widespread recognition as a potential cancer therapy due to its ability to interfere with cancer cell anabolic metabolism. Numerous studies have employed AMPK activators such as metformin and AICAR to demonstrate growth inhibition and increased apoptosis in experimental cancer models.10,17,31-37 In these reports, the growth phenotype was associated with traditional AMPK target regulation, including mTOR inhibition, fatty acid synthase downregulation, and ACC downregulation. In addition, there is increasing evidence that AMPK directly targets p53 to induce apoptosis7,9,38,39 and indirectly suppresses cyclin D1 levels.28,29 Together, these metabolic and cell growth modifications have provided potent anticancer effects in the laboratory setting among a wide array of transformed cell types including breast, prostate, ovarian, lung, and colorectal.17,31,40,41
A recent study presupposed that high metabolic demands from tumors would be sufficient to activate AMPK in situ without chemical induction. 27 Surprisingly, immunohistochemical analysis of primary human breast cancers revealed that AMPK was not phosphorylated in the majority of breast cancer cases, suggesting that dysfunction may exist somewhere within the AMPK activation process. One possibility for this observation is that the upstream AMPK kinase, LKB1, is mutated in breast cancer, although most studies indicate that this is an uncommon event.42,43 Another explanation is that AMPK is unable to be activated due to a phenomenon occurring at the level of AMPK itself; however, neither total AMPK α nor the individual α isoforms were evaluated for protein expression in this benchmark study. 27
To better understand how AMPK dysfunction could lead to cancer initiation and progression, the 2 catalytic isoforms, AMPKα1 and AMPKα2, were evaluated in the normal mammary epithelium as well as primary and experimental human breast cancer samples. The importance of AMPK signaling in normal mammary epithelial tissue was demonstrated using primary HMECs. Both HMEC lines expressed AMPKα1 and AMPKα2 proteins and were sensitive to nutrient restriction. When AMPKα2 was silenced using RNAi, the HMECs failed to inhibit cyclin D1 expression under stress, which may establish a permissive environment for aberrant cell growth and mutations.
Using several primary breast cancer tissue platforms, this study demonstrated for the first time a significant and widespread reduction in AMPKα2 protein expression in primary breast cancer tissue samples. Our findings were further validated by independent laboratory submissions in the Oncomine database, including Glück et al., 44 Richardson et al., 45 and Turashvili et al. 46 breast data sets, all showing that AMPKα2 mRNA expression was consistently reduced in breast cancer compared to normal breast samples. In addition, Kim et al. 47 recently described a comprehensive transcriptome analysis of gastric cancer using multiple analytical algorithms, demonstrating that the AMPKα2 isoform was uniquely suppressed in tumors compared to normal samples. This widespread phenomenon suggests that the observed impairment of AMPK activation in situ 27 may be due, at least in part, to the reduced levels of AMPKα2 expression.
Given that AMPKα2 suppression in primary breast cancer eliminates potential growth regulation mechanisms, we hypothesized that AMPKα2 provides a tumor suppressor–like function. An AMPKα2 re-expression model was employed to test whether AMPKα2 could inhibit the tumorigenic properties of estrogen-dependent and p53 wild-type MCF-7 breast cancer cells. Stable expression of AMPKα2 in MCF-7 cells inhibited proliferation rates moderately in vitro yet significantly in vivo. This growth suppression phenotype was associated with increased apoptosis under nutrient stress conditions as well as in MCF-7 tumor xenografts. Interestingly, attempts to stably express AMPKα2 in other breast cancer lines were unsuccessful due to an acute block in proliferation, which may be attributed to excessive cellular aneuploidy and which conflicts with AMPK’s function in chromosomal segregation fidelity (data not shown), a possibility currently under investigation. The tumor suppressor phenotype caused by the expression of AMPKα2 as demonstrated in these experiments complements an alternative approach demonstrated in a model of prostate cancer, showing that AMPK inactivation using dominant-negative overexpression or stable hairpin inhibition dramatically increased tumorigenicity. 48
In addition, the model presented here suggests that the apoptotic effects of AMPKα2 expression may be most effective in cells harboring wild-type p53. While the 2 AMPK α isoforms are highly homologous, increasing evidence suggests differential functions within the cell.49,50 AMPKα2, but not AMPKα1, may regulate p53-induced apoptosis due to selective AMPKα2-p53 complex formation.8,51 As a result, the apoptotic mechanism proposed in this study integrates metabolic signaling cues through the AMPKα2-p53 signaling axis (Fig. 7). Accordingly, cells that are p53 mutant or null would not be susceptible to AMPKα2-p53 downstream effects. Gene expression analysis within this study demonstrated that the wild-type p53 breast cell line, MCF-7, has a distinct selection for low AMPKα2 expression, whereas p53 mutant lines (T47-D, BT-474, and MDA-MB-468) are permissive of AMPKα2 expression. This observation suggests that the loss of AMPKα2 may be a transforming event and can only be re-expressed without toxicity once p53 function has been ablated. Several studies have shown the inhibition of cyclin D1 transcription independently of p53, an observation supported by our findings.52-55 Repression of p53 with siRNA did not hinder the observed cyclin D1 repression in response to low glucose, suggesting that AMPK is exerting its cyclin D1 growth control mechanism independent of p53 (data not shown). Overall, AMPKα2 activation likely exhibits additional growth restriction in both wild-type and p53 mutant cancers by potently suppressing cyclin D1 expression and by suppression of the mTOR axis (Fig. 7). The mechanism of the downregulation of AMPKα2 is unknown and warrants further investigation. Data from this study demonstrate that AMPKα2 is repressed at the mRNA level, suggesting that epigenetic modifications or RNA degradation control mechanisms may play a significant role.
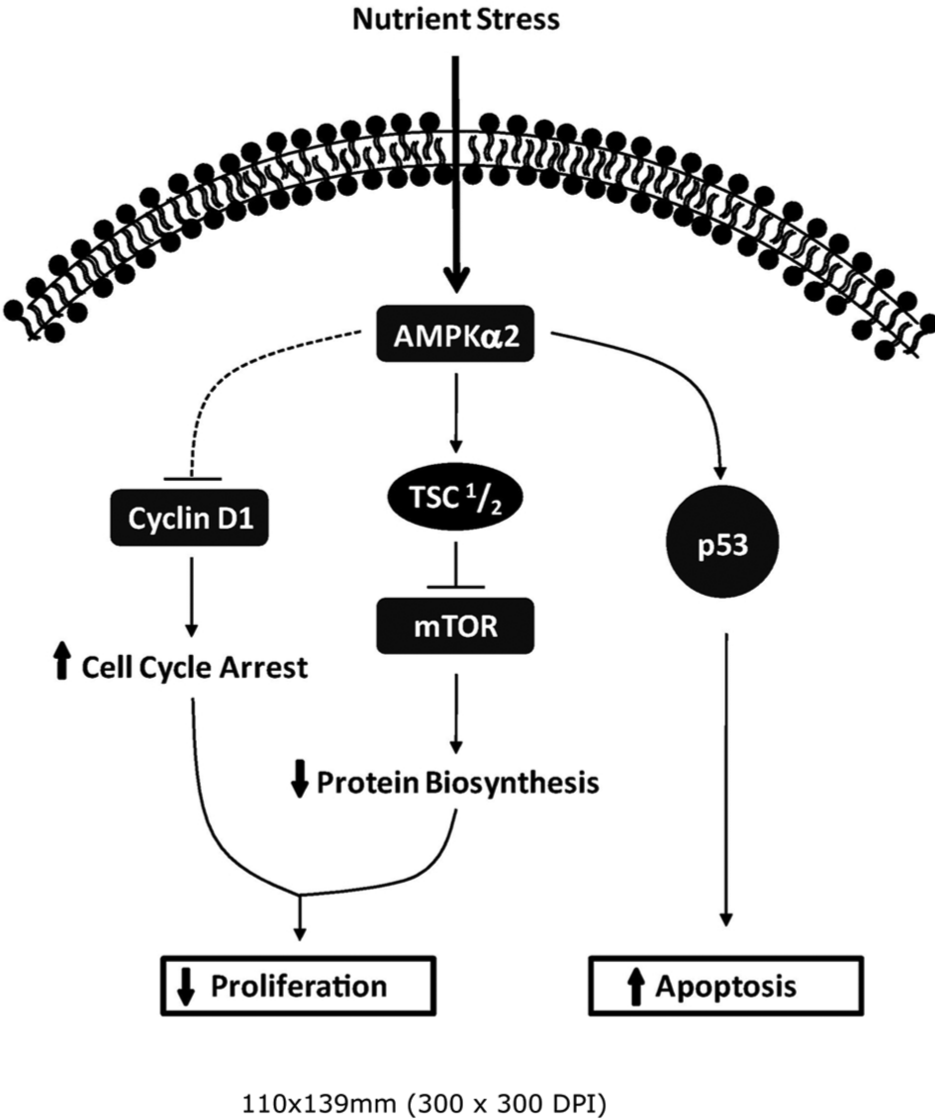
Proposed model of AMPKα2 antitumoral signaling. Re-expression of AMPKα2 in MCF-7 breast cancer cells exerts growth control through 3 distinct signaling pathways: reduced protein biosynthesis through mTOR repression, cell cycle arrest through cyclin D1 downregulation, and apoptosis through p53-dependent mechanisms.
Conclusions
Together, our findings suggest that a delicate balance exists between AMPKα1 and AMPKα2 isoform levels in the normal breast epithelium, and their relative abundance may dictate differential downstream signaling, particularly in p53 and cyclin D1 effectors. Additionally, this work emphasizes the importance of the evaluation of AMPK α isoform–specific expression and signaling events in breast cancer. In this study, low glucose stress induced potent AMPKα2-mediated growth restrictions in both normal mammary epithelial cells and breast cancer cells. The antineoplastic properties of AMPKα2 were further supported in a p53 wild-type, estrogen receptor–positive breast cancer model in which cells also underwent apoptosis more readily than control cells. Combined with our findings that AMPKα2 is suppressed in primary breast cancer, these data implicate the AMPKα2 isoform as a key regulator of proliferation that may serve to inhibit early transformation and progression events in mammary epithelial tissue.
Materials and Methods
Cell lines and reagents
Cell lines were purchased from and maintained according to the American Type Culture Collection (Manassas, VA). Primary HMECs were purchased from Lonza (Walkersville, MD) and were maintained with mammary epithelial growth medium supplemented with a bullet kit. Antibodies targeting AMPKα1 and AMPKα2 were obtained from US Biological (Swampscott, MA); total AMPKα, p-AMPKαThr172, p70S6K, p-p70S6KThr389, cleaved PARP, cyclin D1 (immunohistochemistry), and GFP from Cell Signaling Technology (Beverly, MA); cyclin D1 (immunoblot), p53, and PECAM-1/CD31 from Santa Cruz Biotechnology (Santa Cruz, CA); Ki67 from Dako North America (Carpinteria, CA); ApopTag Plus Peroxidase In Situ Apoptosis Kit from Millipore (Billerica, MA); and β-actin from Abcam (Cambridge, MA).
HMEC RNAi and MCF-7 overexpression transfections
Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used to transfect HMECs with either siLuciferase or siAMPKα2 SMARTpool oligomers (Dharmacon, Lafayette, CO) and MCF-7 cells with either pcDNA3.1/AMPKα2-V5 or pcDNA3.1/GFP vectors. Positively transformed MCF-7 pools were obtained by selecting with 2.0 mg/mL G-418 (Invitrogen).
Cell culture growth assays
Nutritional stress assays were performed by replacing regular growth media (4.5 g/L) with low glucose media (0.45 g/L) 24 hours after plating and allowing cells to grow an additional 18 to 72 hours. Cell growth assays were measured by counting live cells in triplicate or by MTT assays as described previously. 56 Flow cytometry analyses were performed on fixed cells in 40 ug/mL propidium iodide staining solution containing 10 ug/mL RNase A (Invitrogen) for 20 minutes at room temperature. Cell cycle data analysis was performed using ModFit LT software (Verity Software House, Topsham, ME).
SDS-PAGE and immunoblots
Whole cell lysates were harvested and analyzed by Western blot as previously described. 56
Quantitative RT-PCR and Illumina array
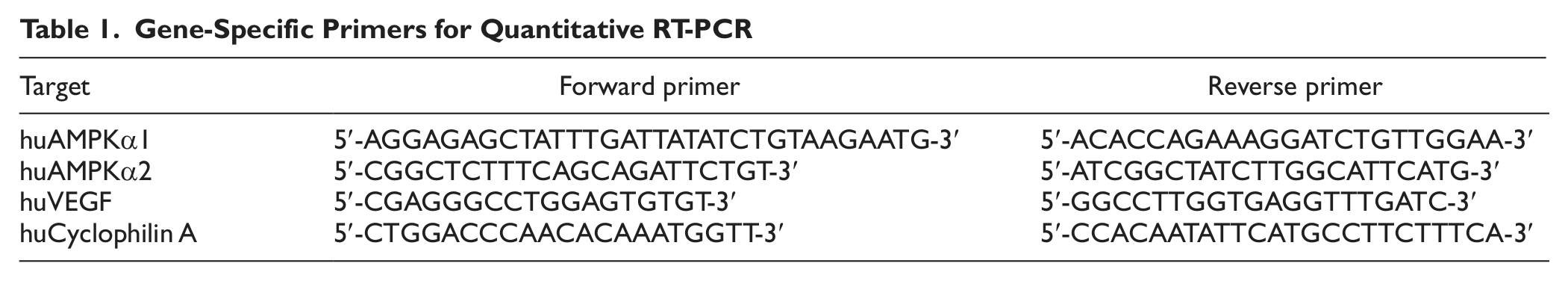
RNA isolation and cDNA synthesis were carried out using the RNeasy mini kit (Qiagen, Valencia, CA) and iScript cDNA synthesis kit (Bio-Rad, Hercules, CA), respectively. Quantitative PCR was then performed with SYBR green fluorescence on a Bio-Rad thermocycler with a MyIQ detection system using gene-specific primers (Table 1). Illumina array (Illumina, Inc. San Diego, CA) services were provided by the Translational Genomics Core at the University of Connecticut Health Center. The array was normalized using a rank invariant model, and only statistically significant signals were used for analysis.
Gene-Specific Primers for Quantitative RT-PCR
Orthotopic xenograft model
Estradiol-17β time-release capsules (0.72 mg/pellet, 90-day release) (Innovative Research of America, Sarasota, FL) were implanted subcutaneously in the anterior dorsal region of each mouse 7 days prior to cell inoculation. Xenografts were established by injecting 8.33 × 105 MCF-7 cells expressing GFP or AMPKα2-V5 suspended in 7 mg/mL BD Matrigel matrix (BD Biosciences, San Jose, CA) subcutaneously in the mammary fat pad of female athymic (nu/nu) mice. Tumor growth was monitored by external caliper measurements over 36 days, and the estimated ellipsoid volume was calculated as width × length × height × 0.52. Xenograft model data are representative of 3 independent experiments showing similar results.
Histological assessment
Paraffin tissue sections were analyzed with immunohistochemistry as described previously. 28 Staining intensity was scored by 3 independent reviewers for the primary human tissue cohort. Samples that displayed no staining within epithelial cell regions were given a score of 0, while those with positive staining were scored a 1, 2, or 3, where 3 was considered the strongest observed staining intensity. Additionally, samples were given a percentage score based on the percentage of tissue displaying each intensity score. Each score was represented as a histoscore in which the percentage score was multiplied by the intensity score, giving a range of 0 to 300. MCF-7 xenograft sections were quantified from images taken at 5 high-power fields/sections using 5 animals per group. Positive staining was calculated using Image Pro Plus software (Media Cybernetics, Silver Spring, MD) or manual identification. Images were taken on a Zeiss Axioplan 2 microscope with AxioCam MRc camera and AxioVision software (Oberkochen, Germany) at 400× magnification.
Statistical analysis
Data from individual experiments were represented as mean ± standard error unless otherwise stated. Statistical comparison of groups was performed using a 2-tailed Student t test or ANOVA test with appropriate tests for equal variances. Statistical significance was defined and indicated as P ≤ 0.05 (*), P ≤ 0.01 (**), or P ≤ 0.001 (***).
Footnotes
Acknowledgements
The authors thank Poornima Hegde, MD, Frank Vumbaca, Nancy Ryan, and Adam Goldstein for acquisition and technical assistance with the primary breast tissue histology samples. In addition, the authors acknowledge the efforts of Roderick Franczak and Katie Sullivan for their technical assistance in sample preparation and evaluation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health/National Cancer Institute (R01 CA064436 to K.P.C.) and the Department of Defense (W81XWH-10-1-0319 to M.M.F.).