
Abstract
Disseminated cancer cells rely on intricate interactions among diverse cell types in the tumor-associated stroma, vasculature, and immune system for survival and growth. Ubiquitous expression of c-Jun N-terminal kinase (jnk) genes in various cell types permits their control of metastasis. In early stages of metastasis, JNKs affect tumor-associated inflammation and angiogenesis as well as tumor cell migration and intravasation. Within the tumor stroma, JNKs are essential for the release of growth factors that promote epithelial-to-mesenchymal transition (EMT) in tumor cells. JNK3, the least ubiquitous isoform, facilitates angiogenesis by increasing endothelial cell migration. Importantly, JNK expression in tumor cells integrates stromal signals to promote tumor cell invasion. However, JNK isoforms differentially regulate migration toward the endothelial barrier. Once tumor cells enter the bloodstream, JNKs increase circulating tumor cell (CTC) survival and homing to tissues. By promoting fibrosis, JNKs improve CTC attachment to the endothelium. Once anchored, JNKs stimulate EMT to facilitate tumor cell extravasation and enhance the secretion of endothelial barrier disrupters. Tumor cells attract barrier-disrupting macrophages by JNK-dependent transcription of macrophage chemoattractant molecules. In the secondary tissue, JNKs are instrumental in the premetastatic niche and stimulate tumor cell proliferation. JNK expression in cancer cells stimulates tissue-remodeling macrophages to improve tumor colonization. However, in T-cells, JNKs alter cytokine production that increases tumor surveillance and inhibits the recruitment of tissue-remodeling macrophages. Therapeutically targeting JNKs for metastatic disease is attractive considering their promotion of metastasis; however, specific JNK tools are needed to determine their definitive actions within the context of the entire metastatic cascade.
Keywords
Introduction
Cancer is a disease affecting millions of people worldwide, with approximately 7.6 million people dying from cancer each year. While primary tumors undoubtedly impair patient health, patients most often die of metastatic disease. It was previously thought that metastasis only occurred in the late stages of tumor progression; however, it is becoming accepted that cells in the primary tumor acquire metastatic potential early. 1 For these reasons, it is of paramount importance to consider the steps of the metastatic cascade early in treatment regimens.
Metastasis is a complex, multistep process whereby tumor cells acquire the characteristics necessary to escape their original environment, survive in the bloodstream, and seed and propagate at distant sites. Dissemination of cancer cells from the primary tumor involves interactions of rogue tumor cells with diverse cell types of the tumor-associated stroma, vasculature, and immune system. During the initial escape from the primary tumor, cancer cells exchange signals with stromal fibroblasts as well as macrophages within the tumor microenvironment to promote motility and invasiveness. In the bloodstream, cancer cells evade detection by circulating immune cells and then interact with distant vascular cells to invade back through the endothelial cell layer. Furthermore, macrophages dispersed ahead of the tumor cells are responsible for constructing the premetastatic niche for disseminated tumor cells to grow. Lastly, interactions between the tumor cells and their new surrounding tissue determine whether these cells remain dormant or resume growth to form macrometastases. Herein, we summarize compelling evidence that implicates c-Jun N-terminal kinase (JNK) proteins in every step of the metastatic cascade, both through their expression in tumor cells and in other interacting cell types to promote the spread of disease.
JNKs were initially described as being highly activated by cellular stresses and were, as such, labeled “stress-activated protein kinases.” 2 Of the 3 jnk genes (jnk1-jnk3), jnk1 and jnk2 are expressed in all tissues; jnk3 is less studied, and its expression pattern is believed to be more limited. Each of the genes encodes several splice variants that result in 10 isoforms with molecular weights ranging from 46 to 54 kDa. Importantly, there is ample literature attempting to describe discrete or even opposing functions of the two ubiquitously expressed JNK genes. For example, loss of jnk2 in mouse embryonic fibroblasts (MEFs) increases cell proliferation, whereas a loss of jnk1 leads to decreased proliferation. 3 To date, these effects are attributed to differential regulation of the mitogenic transcription factor, c-Jun. c-Jun is a part of the AP-1 transcription factor, which is essential for proliferation and cell survival. 4 JNK1 preferentially phosphorylates c-Jun, whereas JNK2 decreases c-Jun stability. Our laboratory has observed similar phenotypic effects in 3-dimensional cultures of primary mammary epithelial cells. In this case, jnk2–/– acini grow at a higher rate than wild-type counterparts, but jnk1–/– acini grow much more slowly (unpublished data). These results highlight the disparate functions of JNK isoforms and show the necessity to study them individually and in combination to properly grasp their impact on biology.
With that being said, ascribing functions to individual JNKs has been complicated by the lack of available pharmacological inhibitors for individual isoforms, as all JNK isoforms are homologous in their kinase domain. As a result, most pharmacological JNK inhibitors affect the activity of all JNK isoforms. Further, some pan-JNK inhibitors target other protein kinases at frequently used concentrations. Fortunately, more specific JNK inhibitors have recently been developed that demonstrate the efficacy of this tool.5,6 Additionally, a JNK2-selective inhibitor was developed that attenuates JNK2-mediated breast cancer cell migration. 7 This new generation of inhibitors, combined with knockdown and knockout studies, will no doubt be instrumental in the study of JNKs and their unique role in tumor progression. For the purposes of this review, the shortcomings of currently available JNK inhibitors should be considered.
JNKs Promote Epithelial-to-Mesenchymal Transition in Tumor Cells
In the first step of the metastatic cascade, rogue cells exhibit an epithelial-to-mesenchymal transition (EMT) phenotype and have the capacity to degrade their underlying extracellular matrix. EMT is the process whereby epithelial cells break free of connections to adjacent cells, lose apical-basal polarity, and acquire invasive potential. The hallmark of EMT is down-regulation of the adherens junction protein of epithelial cells, E-cadherin, and up-regulation of mesenchymal transcription factors. 8 Notably, EMT is common in tissue stem cells and cancer stem cells, begging the question of whether EMT is induced or rather selected for the pressures of the metastatic cascade. 9 If metastasis is driven by cancer stem cells that exist independently of EMT-inducing factors in the environment, these cells could be resistant to treatments that target EMT. The current literature focuses on the induction of EMT in tumor cells, but this issue is especially relevant given the ability of cancer cells to metastasize relatively early in tumor development. 1
Recently, the Davis group reported that JNKs regulate EMT and EMT-associated phenotypes using compound jnk1–/–;jnk2–/– MEFs and conditional jnk1Δ/Δ;jnk2–/– mammary epithelial cells (both will be referred to as JNK deficient).10,11 In MEFs, JNK deficiency, combined with p53–/–, causes mesenchymal-to-epithelial transition (MET), as evidenced by increased E-cadherin, decreased N-cadherin, and decreased colony-forming ability compared to controls. 10 These results support a role for JNKs in promoting EMT and potentially stem cell maintenance. However, in mammary epithelial cells, the JNK-deficient phenotype is contradictory; noncancerous JNK-deficient mammary epithelial cells exhibit an increased in vitro migratory and invasive potential, although EMT markers were not assessed. 11 Our own studies have shown that JNK2 ablation markedly increases tumor cell differentiation, coinciding with decreased EMT marker expression and metastatic capacity in vivo (unpublished data). These conflicting results may be due to disparate systems, especially in comparing nontumor to tumor models, but further examination is indeed warranted. The fact that normal JNK-deficient mammary epithelial cells are more motile than wild-type and jnk2–/– cells suggests that JNK1 may have a predominant role in mammary cell migration. In this case, the inhibition of cell motility by JNK1 may be epistatic to the promotion of cell motility by JNK2.
JNKs are activated by TGFβ in a cascade that requires TGFβ-activated kinase–1. 12 This mechanism is important for TGFβ signaling because of the requirement for JNK-dependent phosphorylation of SMAD3 in its subsequent transcriptional activation. 13 In this report, not only did the phosphorylation of SMAD3 by JNK potentiate SMAD-dependent gene expression, but it also promoted the nuclear localization of SMAD3. This implies that JNKs promote in an EMT-dependent fashion (Fig. 1), as SMAD3 directly transactivates snai 1 and snai 2.14,15 Indeed, studies of mouse tracheal epithelial cells show that jnk1–/– prevents TGFβ-induced EMT. 16 This same study showed that jnk2–/– had no effect, illustrating the differential importance/roles of the 2 genes in these cells.
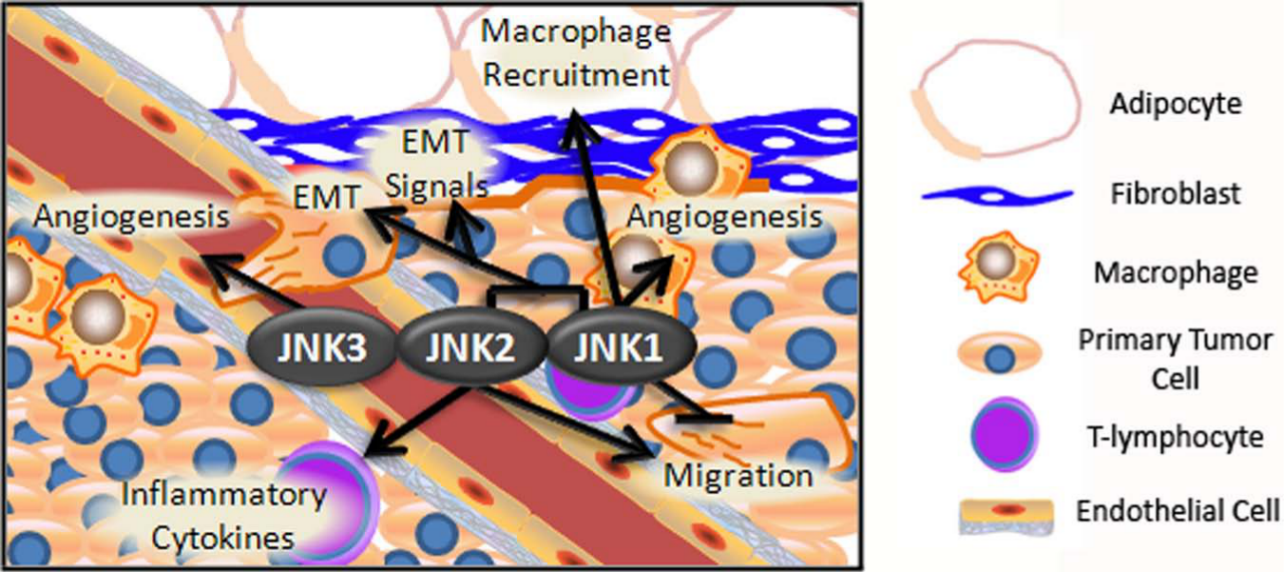
JNK isoforms affect the local invasion of tumor cells. In the early stages of metastasis, JNK isoforms influence metastasis through various cell types. JNK2 enhances inflammation by enhancing the T-cell release of inflammatory cytokines. JNK1 enhances the recruitment of inflammatory macrophages, which secrete VEGF to promote angiogenesis and MMPs to aid tissue remodeling. JNK3 facilitates angiogenesis through facilitating endothelial cell migration. Within the fibroblasts, JNK isoforms are important for the release of growth factors that promote EMT in tumor cells. Within tumor cells, JNK2 promotes migration toward the endothelial barrier, whereas JNK1 inhibits migration. Both JNK1 and JNK2 integrate stromal signals to cause EMT.
JNKs Affect the Recruitment of Inflammatory Cells
In the highly proliferative and hypoxic conditions of a tumor, cells secrete growth factors to attract monocytes to the tumor stroma and promote an inflammatory microenvironment. Monocytes secrete TGFβ, which in turn induces EMT in surrounding epithelial tumor cells.17-20 Removal of this environment drastically reduces tumor growth and progression.21,22 The role of JNK in inflammation is well studied in multiple models. In arthritis, jnk1–/– animals are not protected from inflammation or bone erosion, whereas jnk2–/– animals experience less cartilage damage with no difference in inflammation compared to controls.23,24 In a nonobese diabetic model, JNK2 promotes inflammation by altering cytokine expression by T-cells, 25 whereas JNK1 promotes proinflammatory cytokine expression by liver cells in obesity-driven diabetes. 26 JNK2 also promotes cytokine expression in this model but to a lesser extent. In a further study of the role of JNK1 in obesity-driven diabetes, jnk1–/– was shown to reduce inflammation significantly and prevent macrophage recruitment to adipose tissues; this was attributed to a failure of liver-resident Kupffer cells to produce cytokines 27 (Fig. 1). jnk1–/– similarly reduces inflammation in tobacco smoke–induced lung cancer. 28 In this study, jnk1–/– decreases tumor burden, tumor proliferation, and cytokine expression, including TNFα and IL-6, thus demonstrating the potential influence of inflammation on tumor phenotype. These studies suggest that JNK-dependent inflammatory response influences tumor progression through its induction of EMT in tumor cells and that JNK1 and JNK2 provide unique contributions to inflammation depending on the context in which they are active.
JNKs Facilitate Tissue Remodeling via Stromal Cells
An inflammatory microenvironment also enhances stromal activation and myofibroblast populations, which are important for promoting angiogenesis and tumor cell invasiveness in primary tumors.29-31 TGFβ secretion by macrophages induces myofibroblast expression of matrix metalloproteinases (MMPs), aiding in tumor breakdown of their extracellular matrix. 32 Myofibroblast populations expand in response to the activation of resident fibroblasts; however, others have demonstrated that they arise from the tissue epithelium or endothelium.33,34 The role of JNK in this field has been largely driven by studies using JNK inhibitors with low selectivity, but the results are worth noting for their consistency, intriguing implications, and importance as potential avenues of further study. An early study of lung fibroblasts in culture used CEP-1347 to inhibit JNKs. Treatment of cells with CEP-1347 inhibited TGFβ-induced myofibroblasts. 35 More recently, the same system was studied, using the JNK inhibitor SP600125, and showed similar results, suggesting that JNKs regulate myofibroblast-associated genes, fibronectin (Fn1) and Lef-1. 36 These studies noted JNK activity in isolated fibroblasts, but JNK also promotes myofibroblast-dependent EMT in cancer cells. 37 Conditioned media from MCF10CA1a breast cancer cells, co-cultured with fibroblasts and placed onto MCF10CA1a cells in the presence of SP600125, blocks E-cadherin delocalization to the cytoplasm and decreases cell motility. Additionally, dominant-negative JNK expression and siJNK delivery in tumor cells inhibit the expression of MMP-1 and MMP-9, 2 proteases important for invasiveness.38,39 Collectively, these studies implicate JNK in myofibroblast induction and a broader role of JNK in tumor microenvironment interactions that promote metastasis (Fig. 1).
JNK Isoforms Increase Angiogenesis
In addition to cues that allow for the acquisition of invasiveness, the primary tumor microenvironment provides stimuli to attract prospective metastatic cells toward the vasculature and disseminate them throughout the organism, termed intravasation. The first step in intravasation is the expansion of blood vessels to the tumor microenvironment, a process intimately linked to vascular endothelial growth factor (VEGF) secretion. Macrophages, as noted above, promote blood vessel formation through the secretion of VEGF. These vessels supply oxygen to the metabolically demanding primary tumor. Once tumor cells break contact with adjacent cells and degrade their underlying matrix, the vessels also offer an escape route for rogue tumor cells. JNK1 increases the expression of vegf mRNA through the binding of c-Jun to its promoter.40,41 Additionally, JNK1 dominant-negative expression suppresses the VEGF-induced proliferation and migration of endothelial cells. 42 JNK3 also promotes endothelial cell migration to facilitate angiogenesis 43 (Fig. 1). These effects are further compounded by JNKs’ indirect regulation of angiogenesis through the attraction of macrophages and promotion of myofibroblast populations, which secrete VEGF, as described above.
JNKs Differentially Regulate Migration in Cancer Cells
Besides VEGF secretion, tumor- associated macrophages produce a gradient of epidermal growth factor (EGF) that attracts EGF receptor–expressing tumor cells.44,45 This interaction is supported by a paracrine loop of colony-stimulating factor (CSF). While CSF is expressed by tumor cells, its receptor is expressed by the associated macrophages. 44 Both EGF and CSF are required for tumor cell invasion. Interestingly, macrophages themselves associate with tumors in a gradient fashion, with more macrophages located at the tumor periphery and becoming more scarce near the center. 46 Tumor-associated macrophages directly lure tumor cells toward the vasculature. Our own studies of cell migration using mammary tumor cells in isolation implicate JNK2 as a key player in EGF signaling through its facilitation of EGFR internalization. 47 These studies found that the expression of EGF substrate 8 (EPS8), an actin-interacting protein important in Rac-mediated cell migration,48,49 was greatly inhibited by JNK2. In a complex model, inhibition of EPS8 by JNK2 promotes cancer cell migration. 47 If this mechanism also promotes the expression of CSF by tumor cells, JNK2 could be central to the EGF-CSF loop and tumor cell intravasation. The EGF-CSF paracrine loop is also dependent upon tumor cell CXCR4 expression and can be potentiated by myofibroblast secretion of CXCL12.50,51 In colorectal cancer cells, JNK mediates the CXCL4- CXCL12 axis. 52 CXCL12 treatment of colorectal cancer cells induces Akt phosphorylation in a mechanism that is promoted by JNK. CXCL12 treatment increased cell migration and proliferation. This study implicates JNK not only in tumor cell motility and chemoattraction but also tumor cell survival and proliferation.
JNK1 is also involved in aspects of cytoskeletal organization that affect cell motility. JNK1 phosphorylates paxillin, a protein that is associated with focal adhesions. 53 Phosphorylation of paxillin decreases the migration of several cell types in culture and Schwann cells in vivo53,54 (Fig. 1). Paxillin has been implicated as an important protein for the progression of lung, breast, and bladder cancers.55-58 JNK1 also potentiates migration in the brain through its phosphorylation of microtubule-associated proteins 1B and 2 and stathmin-2.41,59 These are significant findings due to the prevalence of stathmin overexpression in cancers.60-66 In the cases of both paxillin and stathmins, tumor cell migration is regulated by JNK1 targets through their differential effects on microtubule organization.
JNKs Enhance the Survival of Circulating Tumor Cells
Once in the bloodstream, cells are presented with challenges in reaching the secondary site, including surviving hemodynamic shearing forces, escaping immune detection, and avoiding anoikis. 67 Indeed, very few circulating tumor cells (CTCs) survive to form metastases. 68 Sustained Akt phosphorylation in CTCs is important for anchorage-dependent cells to avoid anoikis. 69 One study showed that JNK phosphorylation in response to CXCL12 in colorectal carcinoma cells is necessary for the activation of Akt. 52 In vitro, knockdown of JNK2 in mouse mammary cancer cells decreases the phosphorylation of Akt in response to insulin, HGF, and heregulin-1. 70 Using a BCR-ABL leukemia mouse model, transformed wild-type and jnk1–/– bone marrow were transplanted into wild-type mice. Invasive lymphoblasts from jnk1–/– bone marrow show markedly less infiltration of peripheral organs such as the spleen and liver. This was correlated with increased apoptosis in jnk1–/– lymphoblasts in vitro due to the decreased expression of Bcl-2. 71 These studies indicate that JNK1, in this context, enhances the survival of circulating cancerous cells by preventing the transcription of inhibitors of apoptosis. Thus, JNK1 and JNK2 may cooperate to enhance CTC survival by increasing survival signals and preventing apoptosis (Fig. 2).
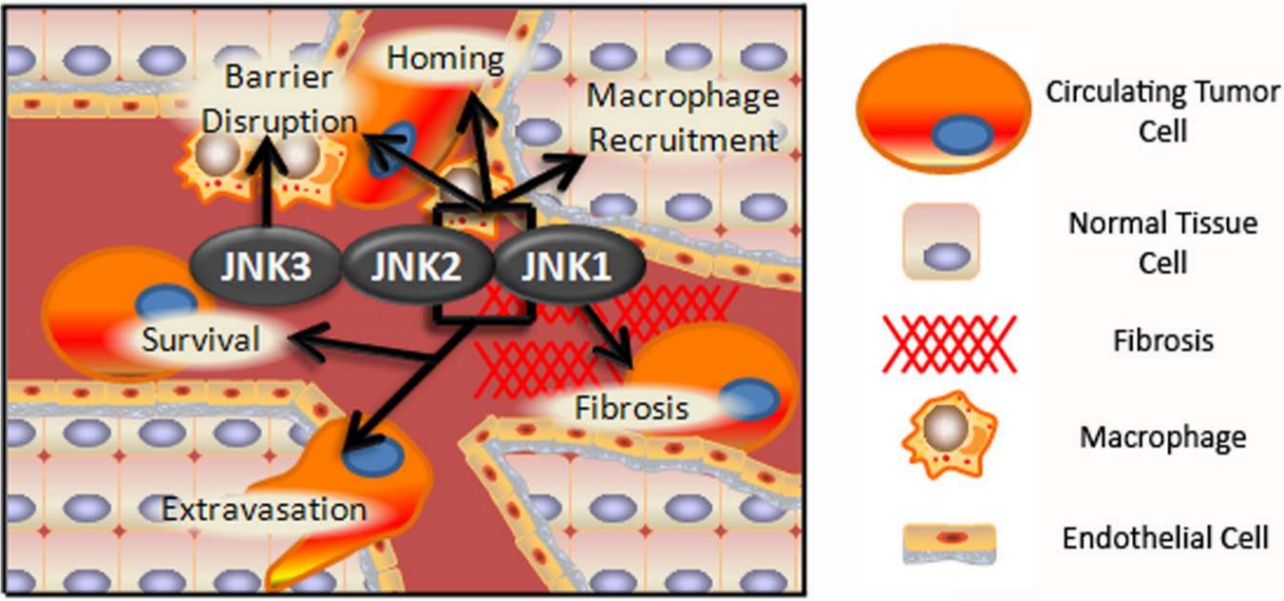
JNK isoforms enhance the survival, homing, and extravasation of CTCs. JNK1 and JNK2 increase the survival of CTCs by inhibiting apoptosis and activating Akt, respectively. JNK activation downstream of CXCR4 contributes to the homing of CTCs to certain tissues. JNK1 is important for fibrosis in tissues being colonized, and both isoforms may contribute to the production of clotting factors in tumor cells and the endothelium, improving CTC attachment to the endothelium. Once anchored, JNK facilitates extravasation by increasing EMT and secretion of endothelial barrier disrupters from tumor cells. Barrier-disrupting macrophages are recruited by the JNK-dependent transcription of macrophage chemoattractant molecules. JNK3 likely facilitates endothelial cell migration to promote barrier disruption.
CTC Homing Is Regulated by JNKs
Although CTCs encounter various tissues while traversing the circulatory system, individual tumor types preferentially metastasize to specific secondary sites. For example, breast cancer cells often metastasize to the bone, lung, lymph nodes, and liver. These differences may not be due to the route that cells take through the bloodstream but rather to chemoattraction by the secondary tissue. Similar to its role in the process of intravasation, CXCR4 is important for the homing of breast cancer cells to CXCL12-expressing tissues, namely the lymph nodes, lung, and bone marrow. 72 As noted above, JNKs mediate the CXCR4-CXCL12 axis and, in this way, facilitate the homing of breast cancer cells to their preferred metastatic niche.
JNKs Improve the Attachment of CTCs to the Endothelium
If CTCs survive long enough to become lodged in the capillary bed of a secondary site, they must firmly attach to the endothelium to extravasate. The local conversion of fibrinogen to fibrin by platelets increases the adhesion of lung carcinoma and melanoma cells to facilitate extravasation and establish lung micrometastases. 73 The transcription of plasminogen activator inhibitor–1 (PAI-1), an essential gene for fibrin deposition, is facilitated by JNK downstream of TGFβ1, with AP-1 directly binding the PAI-1 promoter. 74 Activation of JNK in epithelial or endothelial cells may promote fibrin deposition and increase cell adherence and migration into the secondary site. 75 JNK1, specifically, is important for TGFβ-induced EMT and fibrosis of the lung (Fig. 2). Cooperation between tumor cell EMT and fibrosis of the secondary tissue greatly enhances tumor cell metastasis. 76
Tumor cell binding to various coagulation factors facilitates their arrest in the endothelium. Surprisingly, one study showed that both normal and cancerous mammary epithelial cells produce coagulation factors such as tissue factors and thrombin.77,78 Although no work has specifically shown JNK to be involved in epithelial cell secretion of tissue factors or thrombin, JNK phosphorylation is implicated as an important step in the promotion of their expression in the endothelium.78,79 Additionally, expression of E-selectin, a molecule involved in the adhesion of leukocytes to the endothelium during inflammation, increases after TNFα-mediated JNK stimulation. 80 Through these mechanisms, JNKs facilitate metastasis by increasing the success rate of extravasation by enhancing endothelial attachment.
JNKs Influence Endothelial Disruption to Promote Extravasation
Invasion through the endothelial layer and into the tissue parenchyma requires the disruption of endothelial cell adhesions. COX-2, which is overexpressed in some human breast cancer cells, increases vascular permeability. 81 The tumor promoter TPA induces nonmalignant breast cancer cells to produce COX-2, and this effect is ablated by the SP600125 JNK inhibitor. 82 In lung carcinoma cells, transcriptional inhibition of cox2 is correlated with decreases in JNK phosphorylation, 83 and cell treatment with the SP600125 JNK inhibitor prevents IL-1β–dependent COX-2 transcription. Therefore, JNK activity may facilitate endothelial barrier disruption in CTCs and secondary tissues by enhancing COX-2 expression.
CCL2 is an important chemokine in barrier disruption because it recruits inflammatory macrophages to the site via CCR2 binding. Activation of CCR2 promotes the macrophage secretion of VEGF, 84 another endothelial barrier disrupter. 85 In lung tissue, JNK inhibition by SP600125 diminishes monocyte recruitment by decreasing CCL2 secretion. 86 JNK, through AP-1, activates the transcription of CCL2 in mesangial cells. 87 CCL2 promotes breast cancer cell migration and invasion in vitro 88 and increases the proliferation and migration of prostate cancer cells in vitro. 89 If JNK- dependent CCL2 production occurs in secondary site cells or in CTCs during metastasis, then this will recruit VEGF-secreting macrophages, leading to endothelial barrier disruption and tumor cell extravasation. Considering the role of JNK3 in endothelial cell mobilization during angiogenesis, 43 it may also contribute to barrier disruption during extravasation (Fig. 2).
Tissue Remodeling in the Premetastatic Niche Is JNK Dependent
Macrophages disseminating from the primary tumor arrive at secondary sites ahead of the disseminated tumor cells to establish the premetastatic niche. 90 Along with other immune cells at the secondary site, macrophages initiate tissue remodeling to facilitate tumor cell invasion and to release growth factors from the extracellular matrix that are necessary for cancer cell proliferation. The macrophages and other white blood cells are also affected by tumor-derived growth factors. 90 Co-culture of breast cancer cells with macrophages in vitro induces the invasion of breast cancer cells and corresponds with the rapid phosphorylation of c-Jun. 91 Knockdown of JNK2 by siRNA in breast cancer cells inhibits macrophage-stimulated invasion. Tumor cell and macrophage co-culture also stimulates the secretion of extracellular matrix metalloproteinase inducer (EMMPRIN) and macrophage migration inhibitory factor from the breast cancer cells in a JNK2-dependent manner, in turn promoting MMP secretion by the macrophages 91 (Fig. 3). In vivo, this mechanism may lead to tissue remodeling at the secondary site and formation of the premetastatic niche.
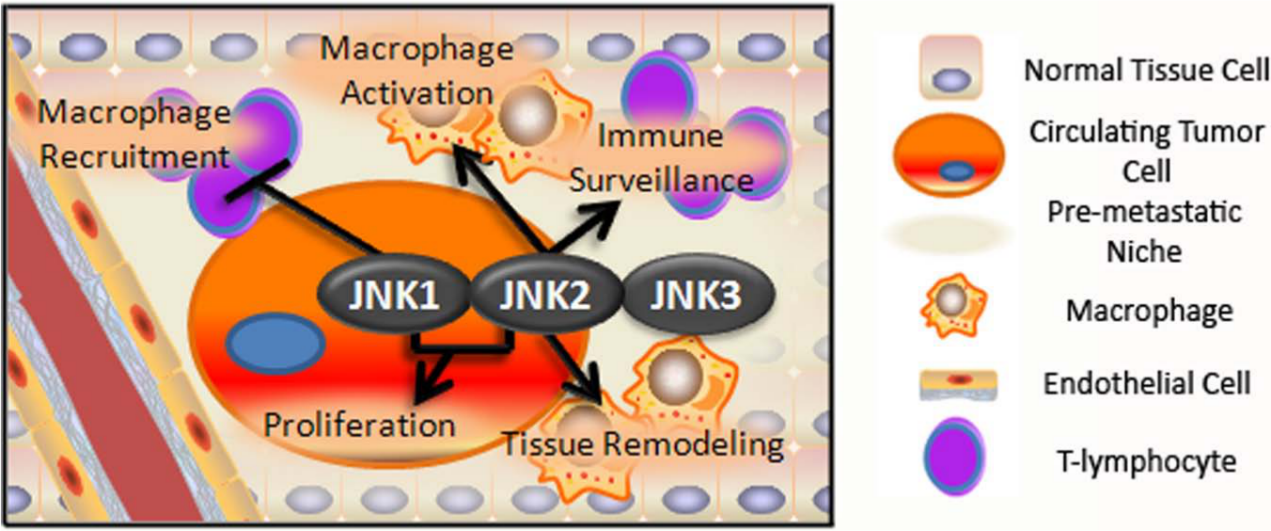
JNK isoforms affect the formation of the premetastatic niche and colonization. JNK2 expression in cancer cells stimulates macrophages to release molecules for tissue remodeling, which improves the chances of tumor cell colonization. JNK2 is necessary in the macrophages to integrate these signals. JNK1 inhibits the recruitment of tissue remodeling macrophages by affecting T-cell differentiation, and JNK2 in T-cells enhances the immune surveillance of seeded tumor cells. JNK2 is necessary for osteoclast differentiation, causing bone resorption that facilitates bone colonization. JNK signaling to promote EMT must be reversed in favor of signaling to promote proliferation.
In the metastatic niche of the bone, cancer cells stimulate osteoblasts to secrete receptor activator of nuclear factor kappa–B ligand (RANKL) that binds to RANK on osteoclasts. RANK induces osteoclasts to resorb bone, releasing growth factors such as TGFβ and IGF.92-94 These growth factors then promote the survival and proliferation of micrometastases, which further promote bone degradation. We have studied integral steps of this “vicious cycle” by using the monocytic cell line, RAW264.7. In this study, we showed that JNK2 is necessary for RANKL-induced differentiation of RAW264.7 cells into osteoclasts (Fig. 3). Indeed, knockdown of JNK2 in RAW264.7 cells reduces RANK expression. JNK2 also increases RANKL expression by mammary tumor cells, but conditioned media from these cells are not sufficient to differentiate RAW264.7 cells. Nevertheless, reduction of JNK2 expression in cancer cells significantly reduces bone metastases and prolongs survival. 70
Necessity of JNK2 in Secondary Tissues
Strikingly, when JNK2 is absent in the host, wild-type cancer cells have reduced metastatic potential to the lung and form indolent bone metastases, indicating that JNK2 is necessary for the growth of seeded tumor cells into overt metastasis. This highlights JNK2’s importance in both the tumor cells and the tissue to be colonized. 70 In contrast to these data, however, expression of MKK4 (the kinase that phosphorylates JNKs and p38 kinases) prevents the growth of lung micrometastatic lesions in a rat model of prostate cancer metastasis. JNK activation was not addressed, but the authors hypothesized that MKK4 allows these cells to undergo stress response signaling, which prevents overt metastasis. 95 Conversely, our data show that JNK2 is necessary to form macrometastases. Later experiments by the same group showed that the expression of MKK4 did not reduce the proliferation or increase the apoptosis of cells in vitro, 96 two processes that JNKs are known to regulate. Experiments by others, however, combined studies of metastasis on MKK4 with MKK6 or MKK7. While MKK4 phosphorylates both p38 and JNKs, MKK6 specifically phosphorylates p38 kinase, and MKK7 specifically phosphorylates JNK. These data showed that the expression of MKK6 provides similar protection against metastasis as MKK4, while MKK7 expression does not. This indicates that the phenotype caused by MKK4 is through its phosphorylation of p38 kinases and not JNKs. 97
JNKs Promote Proliferation in Seeded Tumor Cells
Growth of disseminated tumor cells into macrometastases involves reverting back to an epithelial state, a process termed MET. However, DTCs in secondary tissues can remain dormant for decades before undergoing this reprogramming.98-101 While TGFβ induces EMT, reversal of TGFβ increases proliferation and cohesiveness and re- establishes the self-renewal capacity of metastatic cells.98,99 Although JNKs have a defined role in mediating TGFβ-dependent cell motility and loss of adherens junctions, it is not clear whether TGFβ-dependent JNK signaling increases tumor dormancy or prevents growth programs. 99 Given the importance of JNKs in cell proliferation and that knockdown of JNK2 reduces cancer cell proliferation, it is likely that JNK2 and JNK1 induce the proliferation of cells after EMT reversal. Most likely, loss of TGFβ reduces the availability of JNK substrates that cause EMT, eliminating the competition for phosphorylation by JNK with proliferation substrates, such as c-Jun. Thus, down-regulation of JNKs would not be necessary for the reversal of EMT, leaving them in a position to promote proliferation (Fig. 3).
JNKs Enhance Immune Surveillance at the Metastatic Site
Finally, immune surveillance may play a role during metastatic colonization. Th1 cells convey antitumor responses by secreting IFN-γ, which induces M1-type macrophages and CD8 cytotoxic (CD8+) T-cells. In jnk2–/– lymphocytes, Th1 differentiation is inhibited due to impaired IFN-γ production, 102 indicating that JNK2 promotes immune surveillance. Although most studies show that this occurs at the primary tumor site, immune cells are present in the premetastatic niche, and T-cell differentiation may occur there. Expression of JNK2 in T-cells at the metastatic site would result in decreased metastatic growth. In opposition, Th2 lymphocytes secrete IL-4, which activates M2 macrophages that promote tumor cell growth and tissue remodeling in the primary tumor. Since macrophages are present in the premetastatic niche and tissue remodeling aids in metastatic colonization, Th2 differentiation at the metastatic site would increase metastatic potential. jnk1–/– T-cells show enhanced Th2 differentiation and expression of IL-4 and IL-10, 103 indicating that JNK1 inhibits tissue-remodeling macrophage recruitment at the site of metastasis. This would signify that these genes cooperate in immune cells to monitor and eliminate metastatic cells (Fig. 3).
Problems with Commonly Used JNK Tools
Data presented in this review present a promising future for the potential of JNKs in the treatment and prevention of disease. Mice harboring a single knockout of jnk1 or jnk2 genes do not display altered development or viability, although combined knockout of these genes does result in embryonic lethality. 104 This shows that JNKs are indispensable for development but highlights that JNK1- or JNK2-specific processes may be successful without detriment to organism viability during the early stages of development. Additionally, recent studies by the Davis group show that adult jnk2–/– mice with conditional knockout of jnk1 are viable. 105 Because JNKs mediate processes critical to metastasis and other diseases, they are a promising drug target. Future studies to uncover the potential of JNK inhibitors in treating metastasis should focus on powerful knockout and knockdown models rather than rely on old methods and pan-JNK inhibitors, which show poor selectivity. SP600125 has been used to ascribe general functions to the JNK family. Although this approach has led to important discoveries, its poor JNK selectivity necessitates that these studies be revisited with more specific tools. Additionally, many older studies have relied on molecular weight to discriminate between JNK isoforms in assays utilizing pan-JNK antibodies. This approach has led to potentially erroneous results as ours, and others’ studies with gene knockouts have shown that both JNK1 and JNK2 isoforms contain both 46- and 54-kDa versions, making them indistinguishable from each other using immunoblot techniques.
Differential Roles for JNK Isoforms
The use of effective and specific tools for observing JNK phenotypes becomes especially apparent considering the varied and sometimes opposing functions of JNK isoforms. New functions are constantly reported, and the involvement of JNKs in metastasis is clearly complex. Enhancement of inflammation, migration, and invasion by JNK2, for example, opposes its antimetastatic characteristics in immune cells, where JNK2 promotes the immune surveillance of tumor cells. On the other hand, JNK1-specific data show that it inhibits cancer cell migration, and its inhibition of Th2 differentiation in the premetastatic niche prevents macrophage-dependent tissue remodeling, impeding cell advancement through the metastatic cascade. Conversely, JNK1 promotes EMT, CTC survival, and tissue homing. The importance of specific events in the metastatic cascade with regards to each JNK isoform and tissue type must be considered when determining the prometastatic or antimetastatic capability of JNKs.
Our studies of mammary epithelial tumor cells consistently demonstrate that JNK2 enhances the migration and formation of the premetastatic niche, overall promoting metastasis.47,70 This indicates that the enhancement of immune surveillance does not outweigh these effects or, alternatively, that expression of JNK2 gives tumor cells a mechanism with which to sidestep the immune system. Although unexplored, this could occur through a JNK2-dependent increase in tumor cell binding to platelets, which shields tumor cells from immune detection and increases survival.73,106 Additionally, the importance of JNK2 in remodeling of the premetastatic niche must not be overlooked. The fact that jnk2–/– bone is resistant to the growth of macrometastases indicates a strong influence of this step on overall outcome in mammary tumor progression. In lymphoma, JNK1 is important for fibrosis within secondary tissues, such as the lung, thus facilitating the homing and extravasation of CTCs. However, many studies in epithelial tissue show that loss of jnk1 promotes an aggressive tumor phenotype.107-110 Because lymphoblasts are already in circulation, progression of lymphoma does not necessarily include early steps of metastasis. These studies would, collectively, indicate that the inhibition of tumor cell migration and tissue remodeling are critically important JNK1-mediated steps. Loss of these safeguards results in an overall increased metastatic potential of JNK1-competent tumor cells. Current tools are available to decipher the specific functions of JNKs in these various tissue compartments. These studies could provide much needed clarity on the overall role of JNKs in tumor metastasis.
The lack of studies on JNK3 in tissues other than the brain and testis is disconcerting and probably leads to a neglect of its potential role in important processes of metastasis. Recently, the influence of JNK3 in the circulatory system, including its importance in angiogenesis, was demonstrated. 43 JNK3 is inactivated by promoter methylation in a variety of cancer cell lines, both immune and epithelial in origin. 111 Its function in the prevention of cytokine-mediated apoptosis in pancreatic β-cells has also been noted recently. 112 This mechanism involves Akt2 and, given the established role of Akt in tumor cell survival, could allude to potentially critical JNK3 functions in CTCs. Additional studies to discover the patterns of JNK3 expression in various tissues may find that JNK3 has both compensatory and contradictory functions that affect the ability of JNK1 and JNK2 to carry out their characteristic activities.
Importance of JNK Cross-Talk
It is unclear how the ablation or inhibition of individual isoforms of JNK affects the activity of uninhibited isoforms. For example, JNK2 in jnk1–/– cells necessarily possesses an increased potential to interact with mutual JNK binding partners and alter its function. This leads to a question of whether jnk1–/– phenotypes are driven by acquired functions of JNK2 (and JNK3) or if the functions uncovered are truly JNK1 specific. This calls into question whether JNK inhibitors used to treat metastatic disease would be beneficial or harmful. This question has yet to be satisfactorily answered. Several research groups have attempted to address this concern by assessing JNK activity in single and combined JNK-deficient MEFs, diabetes models, and cancer. MEF studies are indeed interesting, but the model does not allow for the assessment of complex tissue behaviors, and results have already been shown to diverge from cancer and metastasis models specifically. Dual and single knockout models of lung cancer and mammary tumorigenesis, however, show great promise. The potential that JNKs possess tissue- specific effects in cancer is very interesting and important for cancer biology. Clearly, many more model- and tissue-specific studies need to be carried out to unravel the convoluted workings of these proteins in various systems.
JNK Inhibitors for Human Disease
A potent JNK inhibitor is currently in phase II clinical trials from Celgene (Summit, NJ). This drug inhibits fibrosis in mice, and patients in these trials are receiving treatment for autoimmune diseases, pulmonary fibrosis, and discoid lupus. Information is not yet available about the efficacy of this inhibitor or its possible side effects, which is a great concern considering the number of tissues in which JNKs are highly active. Yet with further research into the field, inhibitors of specific JNK isoforms could prevent side effects associated with pan-JNK inhibition. Clinical trials with JNK inhibitors will require careful consideration of the most efficacious treatment approaches with other standard therapies to optimize patient outcomes.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: Dr Van Den Berg’s work was funded by the National Institutes of Health.