
Abstract
Cyclin D1 overexpression is found in more than 50% of human breast cancers and causes mammary cancer in transgenic mice. Dysregulation of cyclin D1 gene expression or function contributes to the loss of normal cell cycle control during tumorigenesis. Recent studies have demonstrated that cyclin D1 conducts additional specific functions to regulate gene expression in the context of local chromatin, promote cellular migration, and promote chromosomal instability. It is anticipated that these additional functions contribute to the pathology associated with dysregulated cyclin D1 abundance. This article discusses evidence that examines the functional roles that cyclin D1 may play in cancer with an emphasis on other cyclin family members that also may contribute to cancer and disease in a similar fashion.
Introduction
The cyclin-dependent kinases (CDKs) are a family of serine/threonine kinases controlling progression through the cell cycle. 1 The regulatory subunits of the CDKs, known as cyclins, form complexes with their catalytic partner to function as checkpoint kinases of specific proteins that regulate progression through the cell cycle. The cyclin-CDK complexes govern a linear progression of events that lead cells from a resting state (G0), growth phase (G1), through DNA replication (S), and finally to cell division (M). Abnormalities that occur in any of the phases initiate a signal that triggers a cell cycle arrest until the issue is resolved. There are some 11 cyclins found in human cells, many having subfamily members (e.g., D-type cyclin D1, D2, and D3). Cyclins partner with associated CDKs and assembly factors to affect their canonical roles in cell cycle checkpoint regulation. Several cyclins exhibit noncanonical roles that may be kinase independent. This review is focused on new and emerging roles for cyclin D1 and includes other cyclins that function in a similar manner.
Cyclin Functions in Cell Migration
Cyclin D1 plays an important role in cell cycle progression through the association with CDK4 and CDK6, which phosphorylate and inactivate the retinoblastoma protein pRb, leading to the expression of a subset of proliferation-associated E2F target genes.2,3 In addition to this canonical pRB-dependent effect in cell cycle progression, cyclin D1 functions in cellular migration, DNA damage response and repair, and chromosome stability.4-8 Metastasis is a major cause of death in cancer patients. Cellular migration is essential for tumor metastasis. Macrophages, fibroblasts, and epithelial cells have enhanced adhesion and reduced migration after depletion of cyclin D1.9-11 The cyclin D1K112E mutant that fails to activate CDK4 or CDK6 does not increase cell migration as the wild-type cyclin D1 protein. This suggests that cyclin D1 induction of cell migration is a CDK-dependent function.10,11 Some CDK4/CDK6 substrates have roles in cell adhesion, cell migration, and cytoskeletal remodeling. Phosphorylation of these substrates such as filamin A 12 and Ral GEF Rgl2 13 by cyclin D1–CDK4 contributes to enhanced cell detachment and motility.
Cyclin D1 binding of p27Kip1 contributes to cellular migration. p27Kip1 has effects on cell migration in either a Rac- or a RhoA-dependent manner through inhibition of RhoA. 14 Introduction of p27Kip1 rescued the cellular migratory defect of cyclin D1−/− cells. Cyclin D1 cannot induce migration following p27Kip1 knockdown. This suggests that cyclin D1 association with p27Kip1 may contribute to cyclin D1 functions in cell migration independent of CDK4/CDK6. 11 In addition, cyclin D1 promotes cellular migration by firstly binding p27Kip1 and thereby inhibiting Rho GTPase activity and secondly by transcriptional upregulation of ROCKII and thrombospondin (TSP-1). The frequent amplification and overexpression of cyclin D1 in cancer cells15,16 and its upregulation by mitogenic growth factors, cytokines, ECM proteins, and other genes, 17 which are important in malignant development, suggest that cyclin D1 may have a central role in mediating the invasion and metastasis of cancer cells by controlling Rho/ROCK signaling and expression of TSP-1. 10
Alternate noncanonical roles for cyclins continue to be discovered; they have important implications for their role in cancer and metastasis. The traditional role for cyclin A2 is in the somatic cell cycle at 2 critical points, when it activates CDK2 at the onset of DNA replication and when it activates CDK1 during G2-M transition. During S phase, cyclin A2 is mostly located in the nucleus, where it regulates the initiation and progression of DNA synthesis. 18 Cyclin A2 localizes to the centrosomes in the cytoplasm, where it binds to the poles of mitotic spindles in a CDK- independent manner. In recent studies, cyclin A2 regulated cytoskeletal organization and cell migration independently of its binding to CDK. 19 Depletion of cyclin A2 causes a change in the distribution of actin filaments and an increase in cell migration. Cyclin A2 interacts with, and activates, RhoA, an actin regulator, which in turn negatively regulates migration. In addition, metastatic cancer cells show less cyclin A2 expression than nonspreading tumor cells. 19
Cyclins in the Regulation of Transcription
As well as having defined roles in cell cycle progression, many cyclins also regulate gene transcription and mRNA processing. Over the last 20 years, a large body of work has implicated cyclin D1 in transcriptional regulation. 20 Cyclin D1 physically associates with more than 30 other transcription factors 20 and regulates the transcriptional activity of estrogen receptor and androgen receptor.21-23 The histone acetyltransferases P/CAF, p300, and AlB1 bind to cyclin D1.23,24 Chromatin immunoprecipitation (ChIP) demonstrated cyclin D1 association within target gene promoters, correlated with deacetylation of histone (H3), in particular at H3 lysine 9. Deacetylation of H3 Lys9 was restored upon the reintroduction of cyclin D1, with concomitant recruitment of HDAC1/HDAC3. 17 Thus, cyclin D1 is recruited in the context of local chromatin to specific target genes.25-27 Cyclin D1 recruitment to genomic DNA was also associated with the shuttling of the histone acetyltransferase p300/CBP to regulate genes governing DNA damage repair signaling. 26 Cyclin D1 was shown to regulate the activity of p300 in a kinase-independent manner. As p300 is regarded as a transcriptional co- integrator, cyclin D1 was proposed as a regulator of gene transcription through co-occupancy with p300 at target DNA binding sites. 26
Protein-coding genes are transcribed by the transcriptional machinery, a multicomplex protein composed of transcription factors (TFIIB, -D, -E, -F, and -H), the Mediator complex, and RNA polymerase II (RNAPII). 28 The Mediator complex is conserved in eukaryotes and bridges the gap between transcription factors and RNAPII.29,30 Mediator is required for the transcription of all yeast RNAPII genes. 31 Several cyclins are involved in the phosphorylation of the largest subunit of RNAPII to regulate transcription: cyclin C–CDK8, cyclin H–CDK7, cyclin T–CDK9, and cyclin K–CDK12 or CDK13. Phosphorylation of RNAPII occurs in the heptapeptide (YS2PTS5PS7) repeats, referred to as the carboxy terminal domain (CTD). 32 A series of phosphorylation events at S2, S5, and S7 of the CTD affects the transcriptional cycle from initiation to elongation and termination.33,34 Cyclin C–CDK8 associates with Mediator proteins Med12 and Med13 to form a subcomplex that interacts with the core Mediator complex to repress activated transcription and does so through phosphorylating the RNAPII CTD and the cyclin H subunit of TFIIH.
The cyclin H–CDK7–Mat1 (CDK-activating kinase [CAK]) complex binds TFIIH and is the principal S5 kinase that eases promoter clearance and enables the transition to transcription initiation, hence providing a direct link between the cell cycle machinery and transcription regulation.35-39 The cyclin H–CDK7–Mat1 complex also interacts directly with transcription factors to regulate their function. Using mouse embryonic fibroblasts (MEFs) and 3T3-L1 cells, phosphorylation of PPARγ by CDK7 blocks lipogenesis. 40
In humans, cyclin T (T1, T2, and T2b) and CDK9 associate to form a complex termed PTEFb, which phosphorylates CTD S2 of RNAPII to regulate productive transcription elongation.41-43 Like cyclin H, the level of cyclin T does not oscillate during the cell cycle, suggesting that these cyclins perform necessary functions that are not cell cycle stage specific.44-47 Cyclin T1 expression is regulated during T-cell activation.48-51 Cyclin T–CDK9 are important regulators of several cellular processes including lymphoid development. 52 Overexpression of cyclin T is sufficient to induce foci and colony formation in NIH 3T3 cells in vitro and induces tumor growth in Nu/Nu mice in vivo. 52 Cyclin T likely contributes to lymphomas derived from B- and T-cell lineages possibly through the inhibition of apoptosis.
Cyclin K binds CDK12 and CDK13, likely in 2 separate complexes to regulate the phosphorylation of S2 and S5 of the RNAPII CTD.53,54 In human cells, depletion of CDK12 results in a marked reduction of CTD S2 phosphorylation. The net effect on gene expression is a downregulation in a modest number of genes; however, these genes have key roles in the DNA damage response (DDR): FANCI, FANCD2, ATR, and BRCA1. 53 Consistent with the role in regulating the DDR, depletion of cyclin K–CDK12 results in sensitivity to DNA damaging agents and increased γ-H2AX foci. 53 Cyclin L (L1 and L2) is closely related to cyclin K, cyclin T1, and cyclin T2. Cyclin L binds CDK11 and interacts with the family of SR splicing proteins to regulate splicing.55-57 Cyclin L is a candidate oncogene in head and neck cancer. 58
Cyclins in the Regulation of Stem Cells
Considerable interest has been given to the hypothesis that stem cells are a central and critical determinant in cancer initiation, maintenance, and recurrence following treatment. The last several years have seen this hypothesis gain in popularity due to a growing body of evidence implicating breast cancer stem cells (BCSCs) as responsible for the origin and maintenance of tumors.59,60 The model proposes that due to the increased longevity of stem cells, they have the propensity to accumulate genetic lesions that could transform the stem cell from a highly controlled and regulated cell to a deregulated abnormal BCSC. The next section will highlight roles for cyclins in stem cells.
Mice lacking cyclin D1 are viable and show deficiencies that are restricted only to a limited set of tissues. Cyclin D1–null animals have hypocellular retinas due to abnormalities in retinal progenitor cell proliferation and retinal cell death.61-63 The abnormality is predicted to be caused by a protracted cell cycle in retinal progenitor cells and early cell cycle exit. 64 Cyclin D1–null animals also display defects in mammary gland development; cyclin D1–null females are defective in lobuloalveolar development during pregnancy and cannot lactate.61,62 Cyclin D1–null mice are resistant to breast cancer induced by Neu and Ras oncogenes. However, animals that are lacking cyclin D1 remain fully sensitive to other oncogenic pathways of the mammary epithelium, driven, for example, by c-Myc or Wnt-1.65-67 Thus, a cyclin D1–targeted therapy could be highly effective in the treatment of human breast cancers in which the primary driver is the Ras oncogenic pathway. The requirement of cyclin D1 in normal mammary gland development appears to be kinase independent. 68 Cyclin D1K112E knockin to the cyclin D1 allele rescued the female mammary gland defect; however, the knockin animals were still resistant to ErbB2-induced tumorigenesis. 69 To explain this puzzling discrepancy, the Hinds laboratory conducted a systematic analysis of the progenitor cell pools in the mammary gland that are dependent on cyclin D1. 68 There are 3 fundamental epithelial cell types in the mammary tissue: luminal, myoepithelial, and alveolar cell types that arise during pregnancy and undergo cell death following the cessation of lactation. All 3 cell types are thought to arise from precursor progenitor cells with self-renewal properties. One type of stem/progenitor cell can be identified with cell surface markers CD24med/CD29HI or CD24+/CD49fHI; a second type is reported to be able to establish a fully functional mammary gland upon transplantation (parity- identified mammary cells: PI-MEC).70-75 An analysis on the cyclin D1KE/KE mutant confirmed that the resistance to ErbB2-driven tumorigenesis is linked to near total absence of the PI mammary cells, making those progenitor cells the likely target for ErbB2-induced tumorigenesis. Cyclin D1 kinase activity is therefore required for mammary progenitor cells’ self-renewal and activity. Recently, acute, conditional ablation of cyclin D1 using a floxed model demonstrated the requirement for cyclin D1 in tumor maintenance. 76 In an MMTV-ErbB2 mammary carcinoma model, deletion of cyclin D1 resulted in reduced tumor cell proliferation and increased cellular senescence, suggesting that the continued presence of cyclin D1 is required to maintain tumor growth in ErbB2-induced mammary carcinomas.
Cyclins appear to have specific and nonredundant roles in stem cell function. Cyclin C was originally cloned from a screen in Saccharomyces cerevisiae conducted to identify factors that could rescue G1 cyclin deficiency.77,78 Subsequently, cyclin C has been shown to promote the progression from G0 quiescence to G1 and does so in part by binding CDK3 to phosphorylate pRB. 79 Recently, an interesting role for cyclin C has been uncovered in the inhibition of hematopoietic stem progenitor cell (HSPC) quiescence. 80 Cyclin C expression was induced upon cytokine activation in HSPCs from human cord blood. siRNA to cyclin C increased the quiescent HSPC population, increased the long-term colony-forming ability, and increased the engraftment capacity.
Cyclin A overexpression correlates with poor prognosis in breast cancer, contributes to prostate cancer invasion and metastasis, and may contribute to colorectal carcinogenesis.81-83 Cyclin A levels increase at S-phase onset. Cyclin A binds to the partners CDK1 and CDK2 and phosphorylates targets that regulate DNA replication (e.g., MCM7).84,85 The cyclin A–CDK complex remains high through mitosis, where it functions to initiate chromosomal condensation and nuclear membrane dissolution.86-88 Cyclin A is redundant in fibroblast cell proliferation but is essential for embryonic and hematopoietic stem cells. 89
Elevated cyclin H is associated with very high-risk gastrointestinal stromal tumors, and reduced or absent cyclin H expression correlates with lower proliferation in B-cell lymphoma.90,91 The transcriptional regulation elicited by the CAK complex (cyclin H–CDK7–Mat1) impacts embryonic stem (ES) cell differentiation. The complex activates CDK1, CDK2, CDK4, and CDK6 through phosphorylation of the T-loop.92-94 Loss of cyclin H function in ES cells induces the differentiation of ES cells and expansion defects of the inner cellular mass in blastocysts. 95 Cyclin H represses ES differentiation possibly through phosphorylation of the negative elongation factor Spt5, an event required for the repressive effect of Spt5 on differentiation. 95 CDK7 phosphorylates Spt5 in vitro, and downregulation of Spt5 leads to the same induction of the differentiation program elicited by the loss of cyclin H.95,96 This is likely transcriptionally regulated since Spt5 regulates RNA processing and transcriptional pausing at sites proximal to the promoter.
Cyclins in DNA Damage and Genomic Instability
Genomic DNA is continually subject to insults by damaging ionizing radiation, chemical carcinogens, and reactive oxygen species generated by cellular metabolism.97,98 In addition, cells are sensitized to errors from DNA replication during S phase. In order to maintain genomic integrity, the cell has several preventative mechanisms relayed through the DDR pathway. Defects in the DDR can lead to genomic instability and cancer. Several cyclin-CDK complexes are implicated in the DDR. 99
Cyclin D1 abundance was shown to determine the DDR, assessed using γ-H2AX and a comet assay. 6 Cyclin D1 was shown to be recruited to the sites of DNA damage, requiring the carboxy terminal exon 5, and to bind directly to RAD51 (a recombinase that drives the homologous recombination process). 6 In a subsequent proteomic screening of cyclin D1, interacting proteins revealed a pool of DNA repair proteins; among them, the most notable was also RAD51. Irradiation of cells stimulated cyclin D1 binding to RAD51 and aided RAD51 recruitment to DNA damage foci in a process that was BRCA2 dependent. 6 This finding was consistent with a prior finding that cyclin D1 bound BRCA1. 100
Reduction of cyclin D1 levels in different types of human cancer cells (mantle cell lymphoma, breast cancer, squamous cell carcinoma, and colorectal cancer) led to the impaired recruitment of RAD51 to the damaged DNA, thus increasing the sensitivity of the cells to radiation. MEFs lacking cyclin D1 showed increased sensitivity to ionizing radiation, which is rescued upon the reintroduction of cyclin D1. This proved to be a kinase-dependent process since the expression of the cyclin D1K112E point mutant and the use of specific CDK4 and CDK6 inhibitors had no effect on the radiation sensitivity. While radiation induces comparable levels of DNA damage in both cyclin D1 control and cyclin D1–depleted cells, the amount of unrepaired DNA after radiation is higher in the cyclin D1–depleted cells.
Cyclin F is unique among the cyclins in that it contains both a cyclin and F-box domain. It does not bind or activate a known CDK and like most cyclins oscillates through the cell cycle. F-box proteins are components of SCF complexes (SKP1–Cullin–F-box); hence, cyclin F acts as a phosphorylation-dependent ubiquitin ligase.101-103 Cyclin F localizes to centrosomes and the nucleus. 104 In the cytoplasmic compartment, cyclin F targets centriolar coiled-coil proteins of 110 kDa (CP110) for degradation. CP110 promotes centrosome duplication; therefore, cyclin F inhibits genomic instability by ensuring that a single centrosome duplication event occurs once per cell cycle.105,106 Additionally, cyclin F degrades nucleolar and spindle-associated protein 1 (NuSAP1) to regulate the correct mitotic spindle architecture. 107 A nuclear role for cyclin F relates to its regulation of RRM2 (ribonucleotide reductase family member 2). 106 RRM2 catalyzes the conversion of ribonucleotides to dNTPs necessary for replication and DNA repair. Failure of cyclin F to degrade RRM2 leads to imbalances in the dNTP pool and increased frequency of genomic mutations. Overexpression of RRM2 leads to lung cancer in mice, and elevated RRM2 in ovarian, colorectal, liver, and breast cancers is associated with poor prognosis.108-112
Downregulation of the cyclin G2 transcript has been linked to various cancers including the thyroid and oral cavity.113,114 Cyclin G1 and cyclin G2 share 53% amino acid identity; however, cyclin G1 lacks the protein-destabilizing PEST domain. 115 The cyclin G1 gene has a p53 binding site and is induced in a p53-dependent manner. 116 The cyclin G2 gene is a transcriptional target of the p53 homolog, p63. 117 Both cyclin G1 and G2 are induced following DNA damage and maintain p53-dependent cell cycle arrest.118,119 Both cyclin G1 and cyclin G2 enhance G2-M checkpoint regulation. In the case of cyclin G1, it may promote or inhibit cell arrest or apoptosis; in the case of cyclin G2, cell cycle arrest is thought to occur through the inhibition of cyclin B1–Cdc2.120-124
Cyclins in the Regulation of Chromosomal Instability
Chromosomal instability (CIN) is a prevalent feature widely shared by cells from solid tumors and is considered a hallmark of cancer.125,126 CIN can be caused by multiple mechanisms and results in an abnormal chromosomal complement. Whether CIN is a cause or a consequence of cancer is a highly debated topic; however, CIN does occur early in cancer development and is associated with poor prognosis. 127 Cyclin D1 is overexpressed in the majority of human breast tumors. Several lines of evidence suggest that, although cyclin D1 is required for tumorigenesis, cyclin D1 conveys a number of cyclin D1 kinase-independent functions. Further, several lines of evidence suggest that the oncogenic function of cyclin D1 may not correlate with its ability to phosphorylate pRB. In this regard, cyclin D1 overexpression does not correlate with pRB phosphorylation or the proliferative marker Ki67 in human breast cancer.128-132
Based on a significant number of publications from our laboratory, and others, we had proposed an alternative mechanism by which cyclin D1 may drive tumorigenesis by inducing CIN. 133 Early studies showed that cyclin D1 did not induce aneuploidy in rat embryonic fibroblasts. 134 A subsequent study on mouse primary hepatocytes showed that transiently expressing cyclin D1 induced abnormal mitosis, accumulation of supernumerary centrosomes, defects in the mitotic spindle, and aneuploidy. 135 Copy number changes in cyclin D1 have been proposed as a biomarker for CIN in bladder cancer. 135 Cyclin D1 gene amplification was only seen in CIN-positive bladder cancer samples and correlated with tumor grade. Our studies, using ChIP of cyclin D1 followed by sequencing (ChIP-Seq), demonstrated that gene regulatory elements bound by cyclin D1 are enriched for genes that govern CIN. 8 Using mammary epithelial-targeted transgenics, the CIN genetic signature 136 was enriched through cyclin D1 overexpression and reduced through the induction of mammary gland–targeted, inducible cyclin D1 antisense. 8 FACS and spectral karyotyping demonstrated that chromosomal duplication was induced by cyclin D1 within 5 cellular divisions. Furthermore, in our studies of 2,254 patients, we showed that cyclin D1 expression correlates with the induction of CIN in luminal B breast cancer. 8 These studies suggest that cyclin D1 promotes CIN as a direct consequence of inducing expression of the mitotic transcriptional program regulated by cyclin D1.
Overexpression of cyclin B1 has been reported in various human tumors, such as colorectal cancer, non–small cell lung cancer, and head and neck squamous cell carcinoma, and its upregulation is closely associated with poor prognosis in breast cancer.137-141 In addition, overexpression of cyclin B1 is related to aneuploidy and high proliferation of human mammary carcinomas. 141 In yeast, overexpression of cyclin B1 and cyclin B2 leads to CIN. 142 In eukaryotes, the maturation/M phase–promoting factor (MPF) regulates entry into M phase.143,144 The MPF complex is composed of cyclin B1 and CDK1 and is sufficient to induce meiotic G2-M–phase transition in immature oocytes and mitosis of somatic cells.145-148 Cyclin B1 mediates chromosome condensation and nuclear envelope dissolution; cyclin B2 mediates Golgi disassembly. The major inhibitor of MPF is protein phosphatase 2A/B55 (PP2A-B55).149,150 MPF suppresses PP2A-B55–dependent inhibition through the Greatwall kinase (Gwl).150,151 Recently, the definition of MPF has been revised to include Gwl as an essential component, at least in frog and starfish oocytes and possibly somatic cells. 152 Depletion of Gwl results in G2 arrest in Drosophila and human somatic cells, and Gwl is required for MPF activity in the oocyte cytoplasm. 152
The abundance of the cyclin E transcript and protein is increased in carcinomas of the lung, gastrointestinal tract, and breast in addition to lymphomas and leukemias.153-159 Increased cyclin E expression occurs in 18% to 22% of breast cancers and has been used as a prognostic marker. 160 A low molecular weight hyperactive version of cyclin E exists in breast cancer and is associated with very poor prognosis.153,161,162 Cyclin E was thought to be an essential cell cycle regulatory protein involved in promoting the G1-S–phase transition. 163 Cyclin E binds its catalytic subunit CDK2 and phosphorylates Rb, in a processive event following cyclin D1–CDK4/6, to release E2F transcription factors that regulate genes involved in S-phase progression. However, a cyclin E knockout model demonstrated that mitotic cell division does not require cyclin E1 and E2, challenging the key requirement for cyclin E in S-phase progression. In addition, development in CDK2-null mice was normal, and null fibroblasts demonstrated a normal cell cycle profile. However, these models do underscore several requirements for cyclin E. Endoreplication of placental trophoblast giant cells and megakaryocytes is severely impaired in cyclin E–null mice.164,165 In addition, null MEFs are resistant to oncogenic transformation, fail to progress from quiescence into S phase, and showed a defect in MCM loading onto replication forks. The absence of these key defects in the CDK2-null mouse may be due to the redundancy offered by cyclin E–CDK1 complexes. Interestingly, the G0-S phase and replication licensing functions of cyclin E were found to be kinase independent. Aberrant cyclin E expression may be caused by gene amplification, defects in the p16–Rb–cyclin D1 signaling axis, or defects in the ubiquitin-mediated degradation pathway.166-168 Increased cyclin E abundance may be a consequence of increased proliferation rates since it often correlates with proliferation indices. 169 However, it may itself act as a molecular driver of transformation and do so through CIN. Induction of cyclin E in rat fibroblasts or human epithelial cells caused aneuploidy. 134 In primary human cells, deregulated cyclin E expression and defective p53 led to increased ploidy and genetic instability. In a lung mouse model, high expression of a degradation-resistant cyclin E targeted to the lung frequently caused dysplasia, multiple lung adenocarcinomas, and tumors exhibiting CIN. 170
Summary
The molecular interplay between the cell cycle, cyclins, and cell function is far from being fully understood. Conceptual advances in the field continue to uncover novel and interesting roles for cyclins in cellular processes that contribute to cancer and disease. In the case of cyclin D1, traditionalists place the protein solely in the RB-E2F signaling axis as a driver of proliferation. However, new functions are emerging that would directly place cyclin D1 as a contributor to cellular transformation and would better explain cyclin D1’s role in oncogenesis, particularly in the fields of stem cell regulation, DDR, and chromosomal stability (Figure 1). Uncovering a breast stem cell population that is dependent on cyclin D1 fits well with previous data that suggest that cyclin D1 is a key inhibitor of differentiation. A contribution of cyclin D1 to enhance DNA repair may protect transformed cells from excessive genomic instability and may help protect breast cancer cells when challenged with DNA-damaging therapies. Cyclin D1 promoting whole-genome chromosome instability is a new discovery. This role for cyclin D1 may be particularly important in the 15% of breast cancers with clonally selected cyclin D1 amplification in which cyclin D1 may be an early driver of oncogenesis through CIN. Future studies should focus on deciphering the key events that cyclins regulate, which instigate and perpetuate cellular transformation.
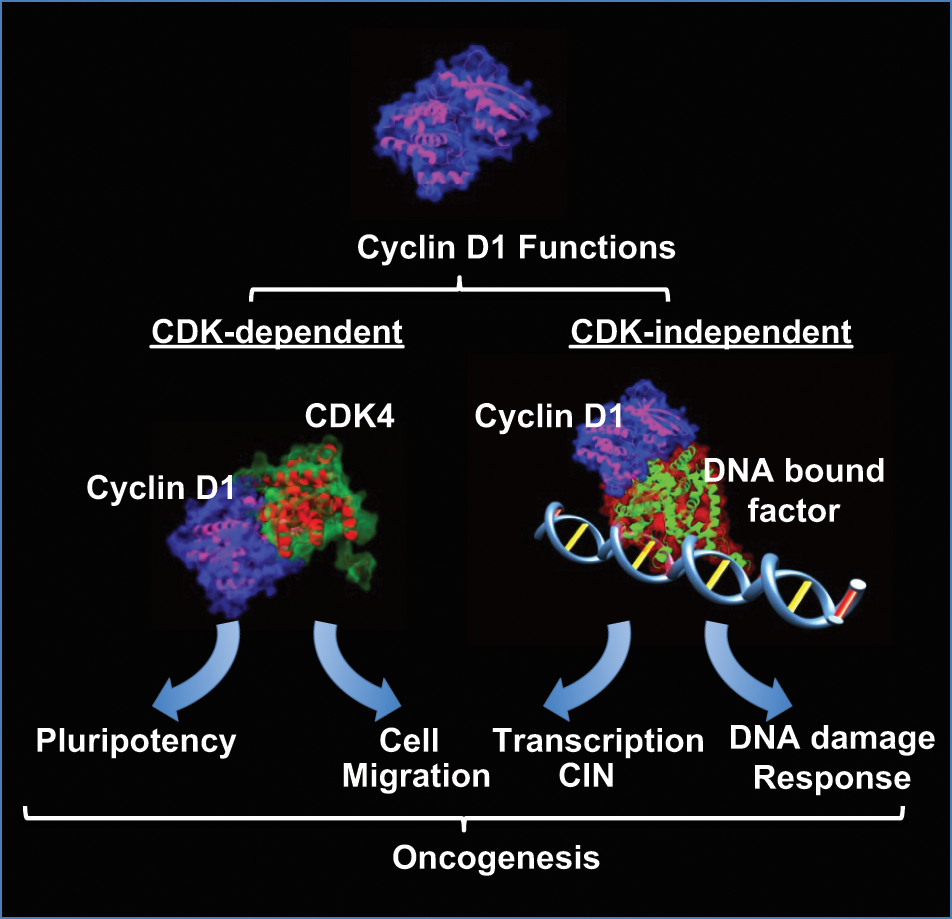
Cyclin D1 forms a holoenzyme through binding CDK4 to elicit kinase-dependent functions that regulate stem cell self-renewal and promote cellular migration. Cyclin D1 functions in a kinase-independent manner to enhance DNA repair and also binds DNA in the context of chromatin to regulate the expression of genes governing CIN. These noncanonical kinase-dependent and -independent functions may contribute to the oncogenic potential of cyclin D1.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This project was funded in part by a grant from the Pennsylvania Department of Health (R.G.P.) and the Breast Cancer Research Foundation (R.G.P.). This project was also funded in part from the Dr. Ralph and Marian C. Falk Medical Research Trust (R.G.P.). The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.