
Abstract
Programmed cell death 4 (Pdcd4), a novel tumor suppressor, inhibits neoplastic transformation and tumor invasion. In this study, the authors found that knockdown of Pdcd4 promoted cell proliferation and up-regulated cyclin D1 expression. Previously, the authors demonstrated that Pdcd4 knockdown activated NF-κB–dependent transcription. Mutations of NF-κB binding sites on the cyclin D1 promoter attenuated the cyclin D1 promoter activity induced by Pdcd4 knockdown. In addition, knockdown of NF-κB/IκB kinase (IKK) α or IKKβ, the kinase regulating NF-κB activation, inhibited cyclin D1 promoter activity and cyclin D1 expression, indicating that up-regulation of cyclin D1 by Pdcd4 knockdown is contributed, at least in part, by NF-κB activation. To investigate the mechanism of how Pdcd4 knockdown activates NF-κB, the authors found that the levels of AKT phosphorylation and AKT kinase activity were increased in the Pdcd4 knockdown cells. Conversely, ectopic expression of Pdcd4 inhibited AKT phosphorylation and cyclin D1 expression, suggesting that Pdcd4 regulates AKT activity and cyclin D1 expression. Furthermore, knockdown of AKT in the Pdcd4 knockdown cells inhibited IKK phosphorylation, NF-κB activation, cyclin D1 promoter activity, and cyclin D1 expression as well as cell proliferation. Taken together, these findings suggest that activation of NF-κB by Pdcd4 knockdown through AKT contributes to the elevated expression of cyclin D1, thus providing new insights into how loss of Pdcd4 expression promotes tumor development.
Introduction
Programmed cell death 4 (Pdcd4) is a novel tumor suppressor that is frequently down-regulated in several types of cancers. In colon cancer tissues, the expression of Pdcd4 is continuously down-regulated in the order of normal-adenoma-carcinoma. 1 Overexpression of pdcd4 cDNA inhibits 12-O-tetraadecanoylphorbol-13-acetate (TPA)–induced transformation in mouse JB6 tumor promotion susceptible cells 2 and tumor phenotype in JB6 transform cells. 3 Recently, Pdcd4 has been shown to suppress tumor cell invasion and intravasation in colon tumor RKO cells4,5 and prostaglandin E2–induced invasion in breast cancer MCF7 cells. 6 Knockdown of Pdcd4 expression promotes invasion in colon HT29 and GEO cells7,8 as well as breast cancer MCF7 and D47T cells. 9 In addition to cell culture systems, Pdcd4 transgenic mice that overexpress Pdcd4 in the epidermis showed significant reduction in 7,12-dimethylbenz(a)anthracene (DMBA)/TPA–induced skin papilloma formation and carcinoma incidence. 10 Conversely, knockout of Pdcd4 in mice resulted in induction of lymphomas with frequent metastasis 11 and an increase in DMBA/TPA–induced skin papilloma formation and carcinoma incidence. 12 These findings suggest that Pdcd4 suppresses carcinogenesis at both promotion and progression stages. Most importantly, a higher Pdcd4 protein level correlates with a good prognosis in lung, 13 colon, 1 and ovarian 14 cancer patients.
Recently, we showed that aerosol delivery of pdcd4 cDNA into a murine lung inhibits cell proliferation and enhances apoptosis in K-ras null mice and AP-1 reporter mice.15,16 Similarly, ectopic expression of Pdcd4 in ovarian cancer cell lines inhibits cell proliferation and cell cycle progression and induces apoptosis. 17 Moreover, immunohistochemical studies showed that expression of Pdcd4 is inversely correlated with the proliferation marker, Ki-76, in the human tissues of intraductal papillary mucinous carcinoma. 18 All of these findings suggest that Pdcd4 is involved in the regulation of cell cycle progression and cell proliferation. However, the mechanisms involved in these processes are still unclear.
AKT (also known as protein kinase B) is one of the most frequently activated protein kinases in human cancer. 19 AKT mediates a large spectrum of cellular functions including cell proliferation, survival, and apoptosis by phosphorylation of its substrates at serine/threonine residues residing in the RXRXXS/T motif. 20 Cyclin D1 is one of the mediators for AKT in regulating cell proliferation. Phosphorylation of cyclin D1 at Thr286 by glycogen synthase kinase 3b (GSK3b) leads to ubiquitin-mediated degradation. 21 AKT-dependent phosphorylation inhibits GSK3b catalytic activity, thus resulting in stabilization of cyclin D1. In addition, AKT can regulate cyclin D1 expression through the NF-κB/IκB kinase (IKK) pathway. 22 The NF-κB family comprises 5 members named p105/p50 (NF-κB1), p100/p52 (NF-κB2), p65 (RelA), c-Rel, and RelB, which form homodimers or heterodimers. 23 NF-κB is mainly activated through IKK. 24 In canonical pathways, IκBs bind with NF-κB complexes in the cytoplasm. Phosphorylation of IκBs by the IKKα/IKKβ complex results in degradation of IκB, leading to nuclear translocation of various NF-κB complexes. In noncanonical pathways, activation of the RelB/p52 NF-κB complex is through processing of p100, a precursor of p52, binding with RelB in the cytoplasm. Upon phosphorylation by IKKα, p100 is processed by proteasome degradation to generate p52, and then the RelB/p52 complex translocates into the nucleus. After translocation into the nucleus, the NF-κB complex activates the expression of NF-κB target genes including cyclin D1. 24
In this study, we demonstrated that knockdown of Pdcd4 promotes cell proliferation and up-regulates cyclin D1 expression, which is regulated, at least in part, through AKT-activated NF-κB.
Results
Knockdown of Pdcd4 expression promotes cell proliferation
To investigate the roles of Pdcd4 in cell proliferation and survival, we stably knocked down Pdcd4 expression in HT29 cells using a lentivirus-mediated system as described previously. 7 The rate of cell proliferation in Pdcd4 knockdown HT29 cells (HT29-shPdcd4) and control cells (HT29-shLacZ) was subsequently analyzed using XTT analysis. As shown in Figure 1A, knocking down Pdcd4 expression in HT29 cells promotes proliferation. The growth rate of HT29-shPdcd4 cells at day 4 and day 5 was approximately 30% to 40% faster than that of HT29-shLacZ cells. Next, we determined whether the increased proliferation in Pdcd4 knockdown cells is due to the reduced cell apoptosis. Cells were stained with annexin V and propidium iodide and measured using fluorescence-activated cell sorting (FACS) analysis. Knockdown of Pdcd4 did not change apoptosis as compared to control cells (2.73 v. 2.67) (Fig. 1B). In addition, the population of viable cells was similar in HT29-shPdcd4 and HT29-shLacZ cells (87.51 v. 89.03) (Fig. 1B). Last, to examine the effects of Pdcd4 knockdown on cell cycle progression, cell cycle distribution was examined using FACS analysis. Compared to HT29-shLacZ cells, HT29-shPdcd4 cells showed a reduction in the percentage of cells in the G1 phase (39.19% v. 30.95%) and an increase in the percentage of cells in the G2/M phase (8.85% v. 17.02%) (Fig. 1C). Because the apoptosis assays (Fig. 1B) indicated that the populations of viable and apoptotic cells were similar in HT29-shLacZ and HT29-shPdcd4 cells, the increase in cell number in the G2/M phase of the HT29-shPdcd4 cells is not likely due to cell growth arrest. It is possible that a fast G1 phase progression of Pdcd4 knockdown cells results in an accumulation of cells in the G2/M phase. Pdcd4 knockdown did not affect the percentage of cells in the S phase and sub-G1 phase. These results demonstrated that knockdown of Pdcd4 increases the rate of cell proliferation but does not decrease the rate of cell death.
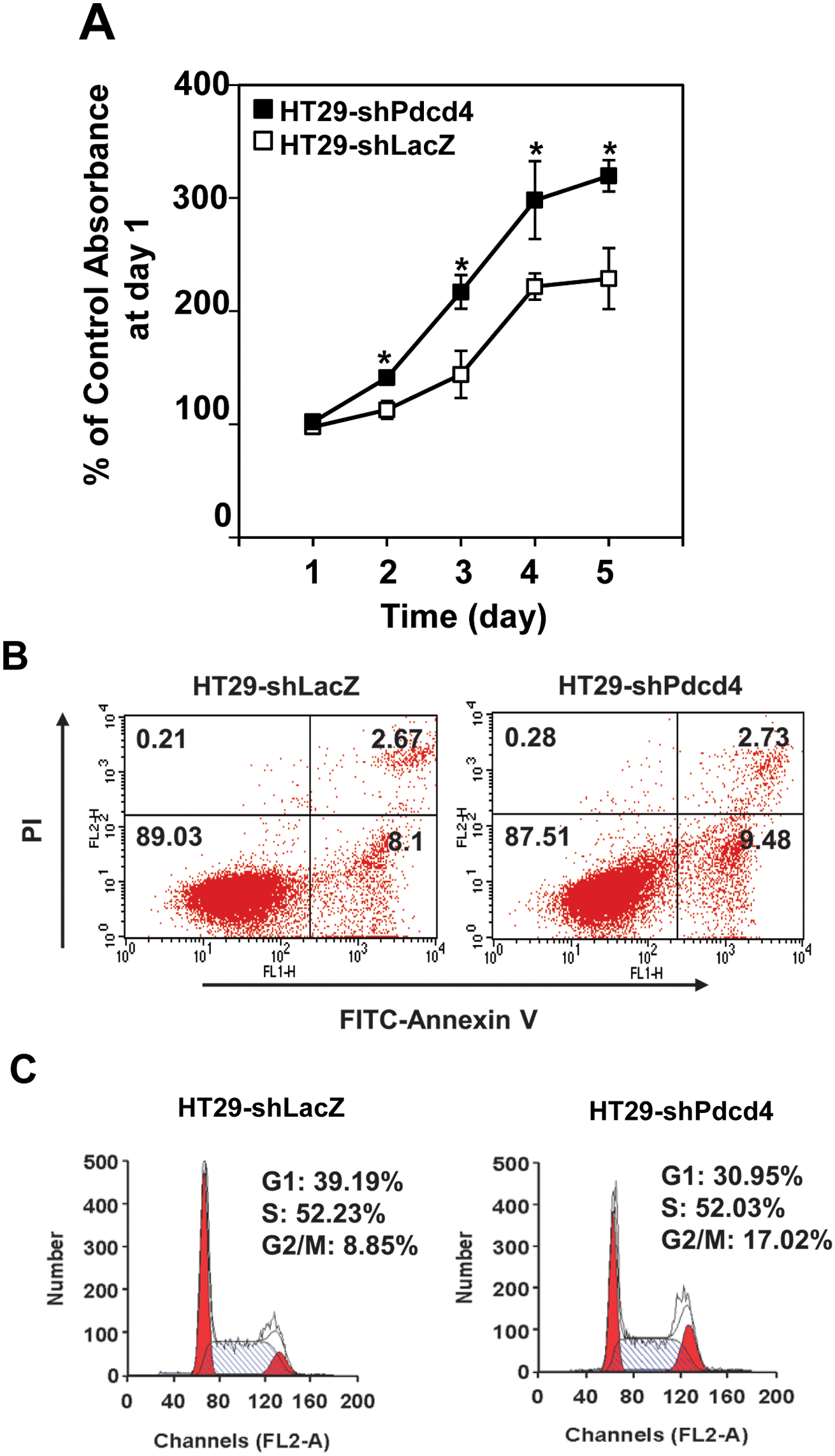
Knockdown of Pdcd4 promotes cell proliferation. (
Knockdown of Pdcd4 up-regulates cyclin D1 expression
The results of cell cycle distribution analysis (Fig. 1C) suggested that knockdown of Pdcd4 shortened the G1 phase progression. It has been known that overexpression of cyclin D1 in cultured cells shortens the G1 phase and increases cell proliferation. 25 Cyclin D1 promotes G1 phase progression through forming active complexes with cyclin-dependent kinase (CDK) 4 and 6, resulting in the activation of genes for G1-S phase transition. 26 To test whether knockdown of Pdcd4 up-regulates cyclin D1 expression, the protein and mRNA levels of cyclin D1 in both HT29-shLacZ and HT29-shPdcd4 cells were examined. As shown in Figure 2A, the cyclin D1 protein level in HT29-shPdcd4 cells was about 21-fold higher than that in HT29-shLacZ cells. Similarly, the cyclin D1 mRNA level was increased approximately 7-fold in HT29-shPdcd4 cells compared to that in HT29-shLacZ cells (Fig. 2B). These results indicate that knockdown of Pdcd4 induces cyclin D1 expression.
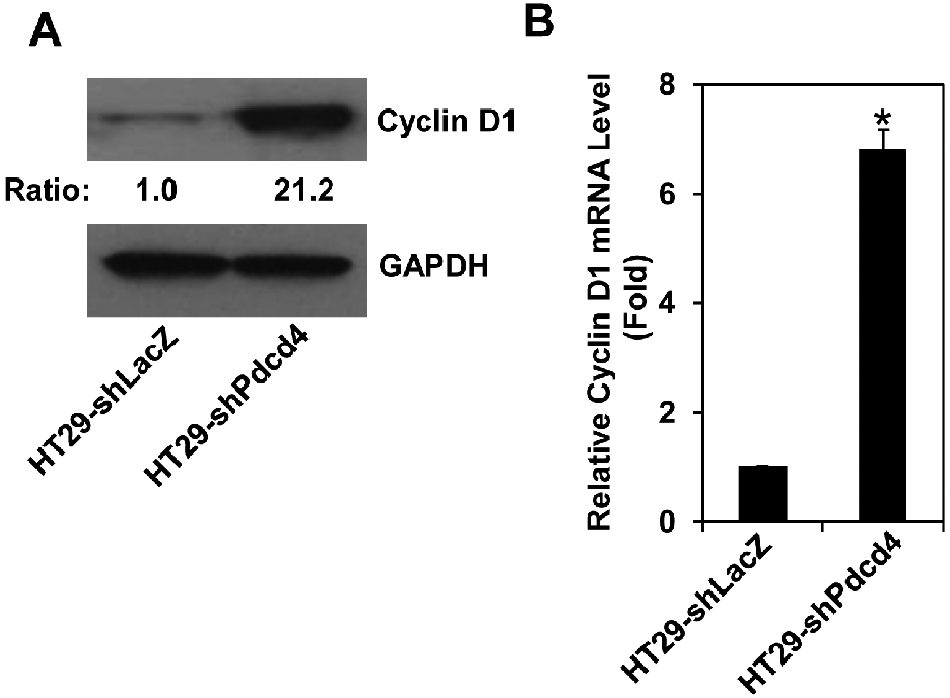
Knockdown of Pdcd4 up-regulates cyclin D1 expression. (
NF-κB regulates cyclin D1 expression in Pdcd4 knockdown cells
The NF-κB complex has been known to play an essential role in the regulation of cyclin D1 expression. 26 Previously, we have demonstrated that knockdown of Pdcd4 activates NF-κB–dependent transcription. 7 To examine whether Pdcd4 knockdown activated NF-κB enhances cyclin D1 expression, we transfected cyclin D1 promoter luciferase reporter plasmid (D1) into HT29-shLacZ and HT29-shPdcd4 cells. The luciferase activity was approximately 32-fold higher in HT29-shPdcd4 cells than that in HT29-shLacZ cells (Fig. 3A, D1), indicating that the promoter activity of cyclin D1 is dramatically activated by Pdcd4 knockdown. The D1 construct comprises a region of the cyclin D1 promoter from –920 bp to +106 bp (relative to the transcription start site) fused with a luciferase reporter, which contains 3 potential NF-κB binding sites located at –858 bp, –749 bp, and –39 bp. 27 To test whether NF-κB binding sites mediate the activation of cyclin D1 promoter in Pdcd4 knockdown cells, the potential NF-κB binding sites were mutated and transfected into HT29-shLacZ and HT29-shPdcd4 cells. All of the mutated cyclin D1 constructs showed a similar basal level of promoter activity in the HT29-shLacZ cells (Fig. 3A, open bars). The D1-858m and D1-749m constructs, in which the NF-κB binding site at –858 bp and –749 bp was mutated, respectively, exhibited approximately 30% lower promoter activity than wild-type (D1) in HT29-shPdcd4 cells. The D1-39m construct, which the NF-κB binding site at –39 bp was mutated, showed approximately 50% promoter activity compared to the D1 construct in the HT29-shPdcd4 cells. When all 3 potential NF-κB binding sites were mutated (D1-m3), the D1-m3 construct exhibited a similar promoter activity in HT29-shLacZ and HT29-shPdcd4 cells, indicating that mutation of these 3 NF-κB binding sites abolished the cyclin D1 promoter activity induced by Pdcd4 knockdown. These results suggest that NF-κB binding sites mediate the cyclin D1 promoter activity in the HT29-shPdcd4 cells.
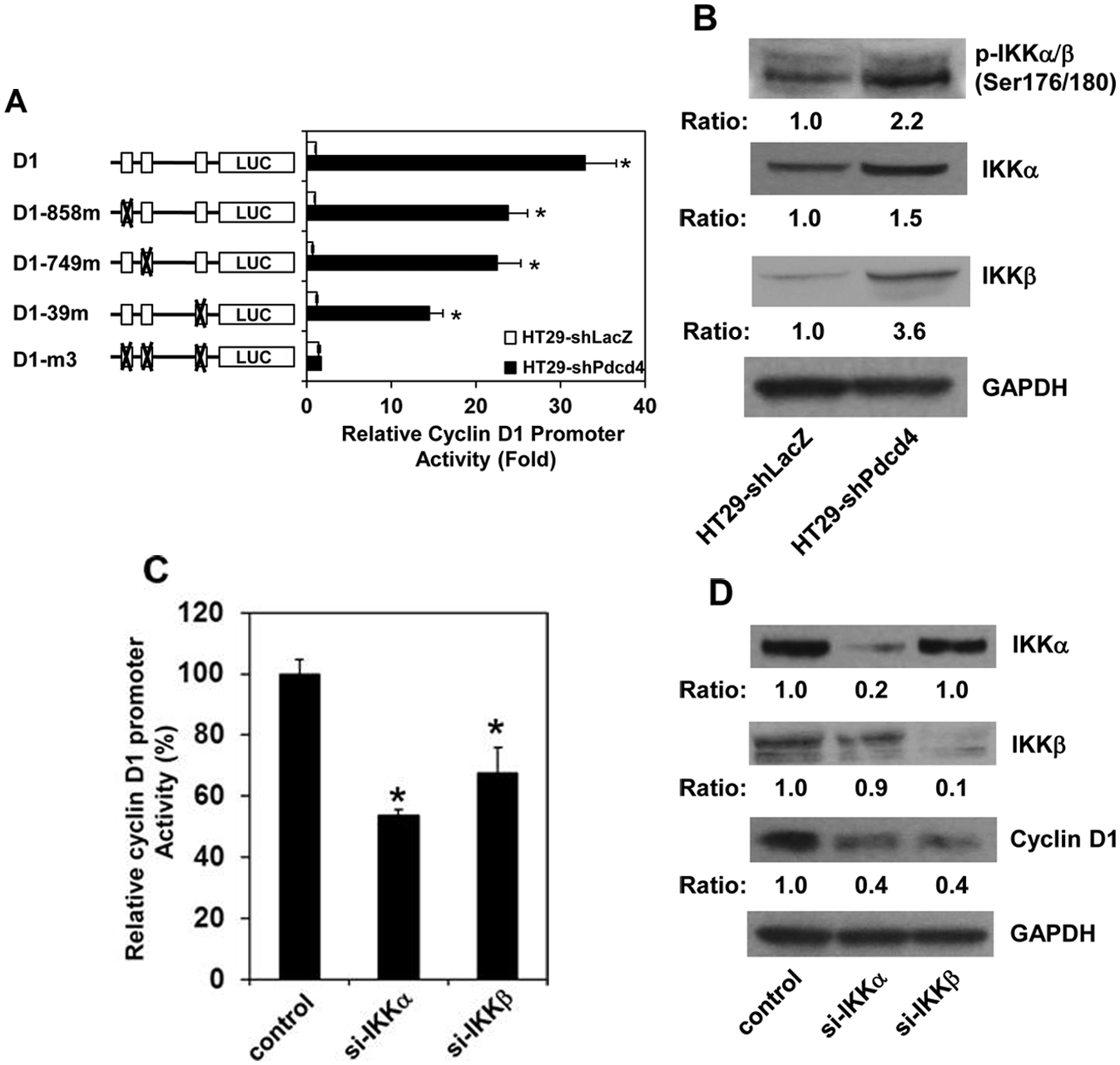
Phosphorylation of IKK activates NF-κB to up-regulate cyclin D1 in Pdcd4 knockdown cells. (
IKKα and IKKβ, 2 major kinases for activating NF-κB, phosphorylate IκB and subsequently lead to translocation of NF-κB into the nucleus. To determine whether IKKα and/or IKKβ play an essential role in activating NF-κB in the Pdcd4 knockdown cells, the phosphorylation status of IKKα/β was first examined in HT29-shLacZ and HT29-shPdcd4 cells. The level of phospho-IKKα/β in HT29-shPdcd4 cells was approximately 2.2-fold higher than that in HT29-shLacZ cells (Fig. 3B). Interestingly, the total proteins of IKKα and IKKβ also showed approximately 1.5- and 3.6-fold higher in HT29-shPdcd4 cells than HT29-shLacZ cells, respectively. Next, we determined whether knockdown of IKKα or IKKβ reverses the cyclin D1 promoter activity induced by Pdcd4 knockdown. HT29-shPdcd4 cells were transfected with IKKα siRNA or IKKβ siRNA. Two days after transfection, these cells were further transfected with the cyclin D1 promoter reporter (D1). As shown in Figure 3C, the cyclin D1 promoter activity of cells transfected with IKKα siRNA or IKKβ siRNA was approximately 50% and 65% of that in control cells, respectively. To further confirm that knockdown of IKKs alters cyclin D1 expression, the HT29-shPdcd4 cells were transfected with IKKα siRNA or IKKβ siRNA. Four days after transfection, the cyclin D1 protein levels were determined. Transfection of IKKα siRNA and IKKββ siRNA HT29 shPdcd4 cells reduced the IKKα and IKKβ protein level to 20% and 10% of that in control cells (control), respectively. In agreement with the results of cyclin D1 promoter activity assays, the cyclin D1 protein in IKKα (si-IKKα) or IKKβ (si-IKKC) knockdown cells is approximately 40% of that in control cells (Fig. 3D). Taken together, these findings suggest that activation of NF-κB by IKKs enhances cyclin D1 expression in the Pdcd4 knockdown cells.
Knockdown of Pdcd4 activates AKT
The AKT signaling pathway plays an important role in regulating cell proliferation, survival, and apoptosis by activating a set of transcription factors including NF-κB.28,29 To test whether knockdown of Pdcd4 in colon HT29 cells activates AKT, the phosphorylation status of AKT in both HT29-shLacZ and HT29-shPdcd4 cells was examined. The level of phospho-AKT (Ser473) in HT29-shPdcd4 cells is approximately 3.8-fold higher than that in HT29-shLacZ cells (Fig. 4A), whereas the levels of total AKT in both HT29-shPdcd4 and HT29-shLacZ cells were similar. Interestingly, the level of phospho-AKT (Thr308) was undetectable in either HT29-shLacZ or HT29-shPdcd4 cells (data not shown). This finding raised the question of whether phosphorylation of AKT at Ser473 is sufficient to activate AKT kinase activity. To test this, the AKT kinase assays were performed using phospho-AKT (Ser473) antibody to precipitate the active form of AKT. The kinase activity of antibody-bound AKT was then assayed by adding the AKT substrate, a recombinant GSK3a/b fusion protein. AKT kinase activity in HT29-shPdcd4 cells was approximately 20.2-fold higher than that in HT29-shLacZ cells, whereas the levels of the total AKT in the cell lysates used to immunoprecipitate phospho-AKT were similar (Fig. 4B). These findings suggest that Pdcd4 knockdown increases AKT phosphorylation and AKT kinase activity.
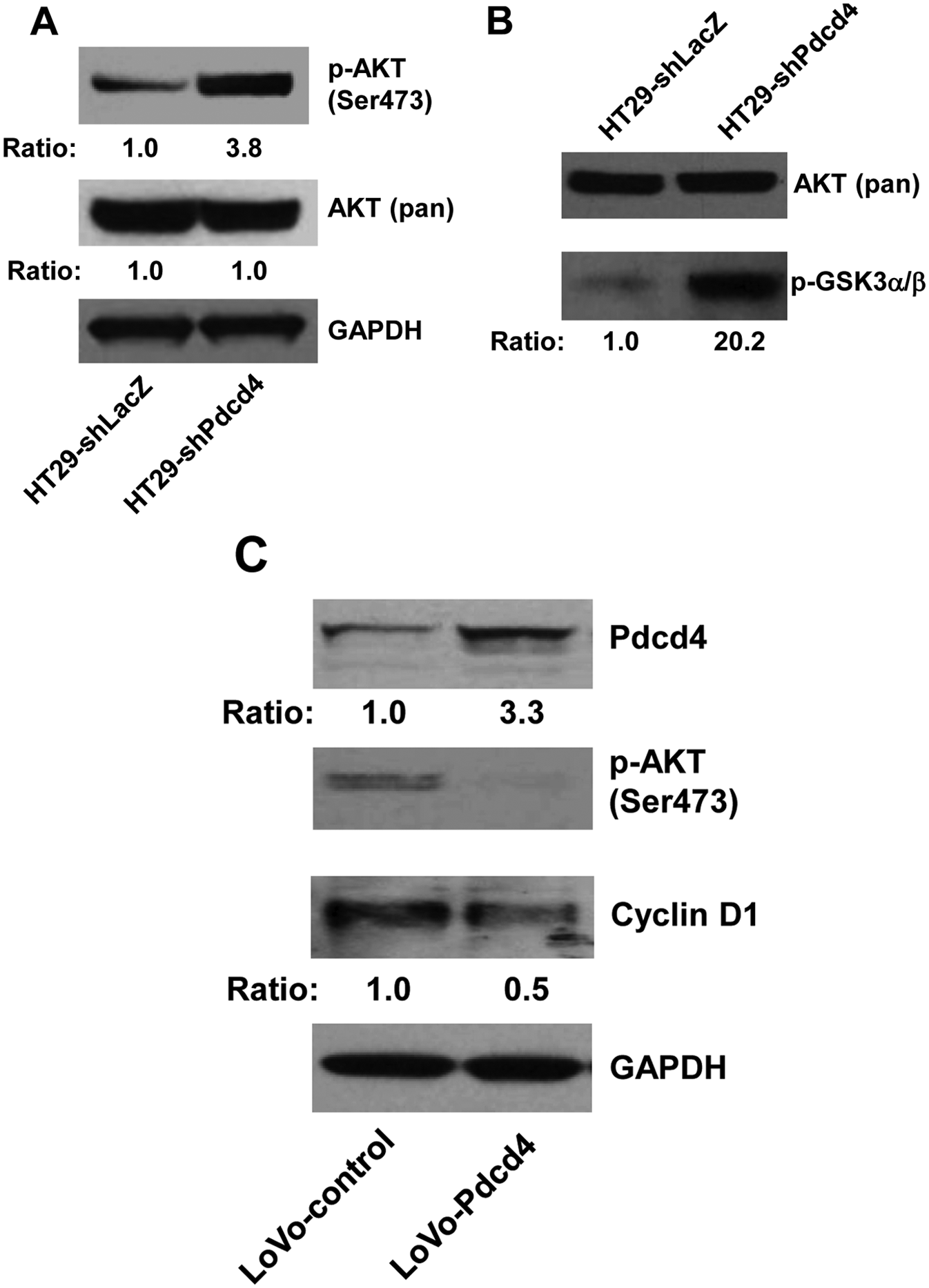
Pdcd4 regulates AKT activation. (
To further confirm that Pdcd4 regulates AKT activity, Pdcd4 was overexpressed in colon LoVo cells using the lentivirus expression system. 8 The Pdcd4-expressing cells (LoVo-Pdcd4) exhibited an approximately 3.3-fold higher Pdcd4 protein level than the cells transduced with lentivirus-empty vector (LoVo-control) (Fig. 4C). The phosphorylation of AKT at Ser473 was dramatically inhibited by overexpressing Pdcd4 (Fig. 4C). In addition, the protein level of cyclin D1 in LoVo-Pdcd4 cells was approximately 50% suppressed by Pdcd4 (Fig. 4C). These findings collectively suggest that Pdcd4 regulates cyclin D1 expression through regulating AKT activation.
GSK3b is involved in regulating cyclin D1 expression in HT29-shPdcd4 cells
It has been known that AKT also can regulate cyclin D1 expression by inhibiting GSK3b activity through phosphorylation of GSK3b, which inhibits proteasome degradation of cyclin D1. 30 We have demonstrated that knockdown of Pdcd4 activates AKT (Fig. 4). Therefore, it is logical to examine whether inhibition of GSK3b activity contributes to the stabilization of cyclin D1 in Pdcd4 knockdown cells. The level of phospho-GSK3b was approximately 2.1-fold higher in HT29-shPdcd4 cells than that in HT29-shLacZ cells, whereas the total GSK3b protein level was similar between HT29-shLacZ and HT29-shPdcd4 cells (Fig. 5A), suggesting that the activity of GSK3b was inhibited in the Pdcd4 knockdown cells. In addition, we found that treatment with MG132 increased the cyclin D1 protein level approximately 9- and 2-fold in the HT29-shLacZ cells and HT29-Pdcd4 cells, respectively (Fig. 5B), indicating that stabilization of cyclin D1 by inhibiting proteasome activity plays a role in up-regulating cyclin D1 in both control cells and Pdcd4 knockdown cells. These results imply that inhibition of GSK3b activity by Pdcd4 knockdown contributes to the elevated cyclin D1 level.
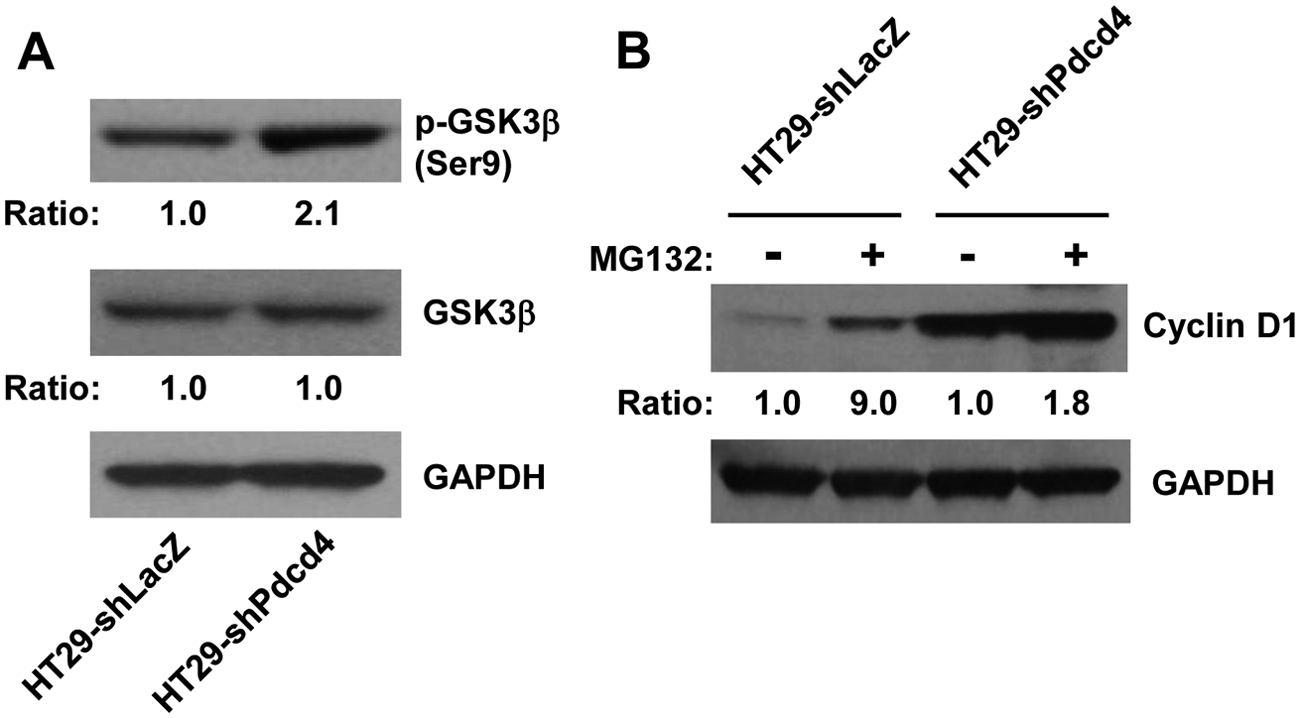
GSK3b is involved in the up-regulation of cyclin D1 in Pdcd4 knockdown cells. (
Knockdown of AKT reverses cyclin D1 expression, NF-κB activation, and cell proliferation induced by Pdcd4 knockdown
To determine the causal role of AKT activation in increasing cell proliferation and cyclin D1 expression in Pdcd4 knockdown cells, we knocked down AKT expression in HT29-shPdcd4 cells using akt siRNA. The levels of total AKT and phospho-AKT in HT29-shPdcd4 cells transfected with akt siRNA (si-AKT) were about 10% of those in cells transfected with scramble siRNA (control), as shown in Figure 6A. Knockdown of AKT in HT29-shPdcd4 cells led to the decreased protein level of cyclin D1, phospho-IKKα/β, and phospho-GSK3β as well as the reduced level of cyclin D1 mRNA (Fig. 6A and 6B), suggesting that AKT contributes to the elevated cyclin D1 expression in HT29-shPdcd4 cells. However, knockdown of AKT did not alter the total protein levels of IKKα and IKKβ (Fig. 6A), suggesting that IKKα and IKKβ expressions are not dependent on AKT in HT29-shPdcd4 cells. In addition, the cyclin D1 promoter activity was also inhibited approximately 50% by AKT knockdown, as shown in Figure 6C. To demonstrate that AKT regulates NF-κB activation, the NF-κB luciferase reporter was transfected into control and si-AKT cells. The NF-κB–dependent transcription was about 40% lower in si-AKT cells than that in control cells (Fig. 6D), suggesting that AKT knockdown inhibits NF-κB activation. Last, cell proliferation in control and si-AKT cells was measured using colorimetric XTT analysis. The si-AKT cells showed slower proliferation compared to the control cells (Fig. 6E). The rate of cell proliferation at day 5 and day 6 was approximately 20% to 30% lower in si-AKT cells than that in control cells, indicating that knockdown of AKT reverses the cell proliferation induced by Pdcd4 knockdown.
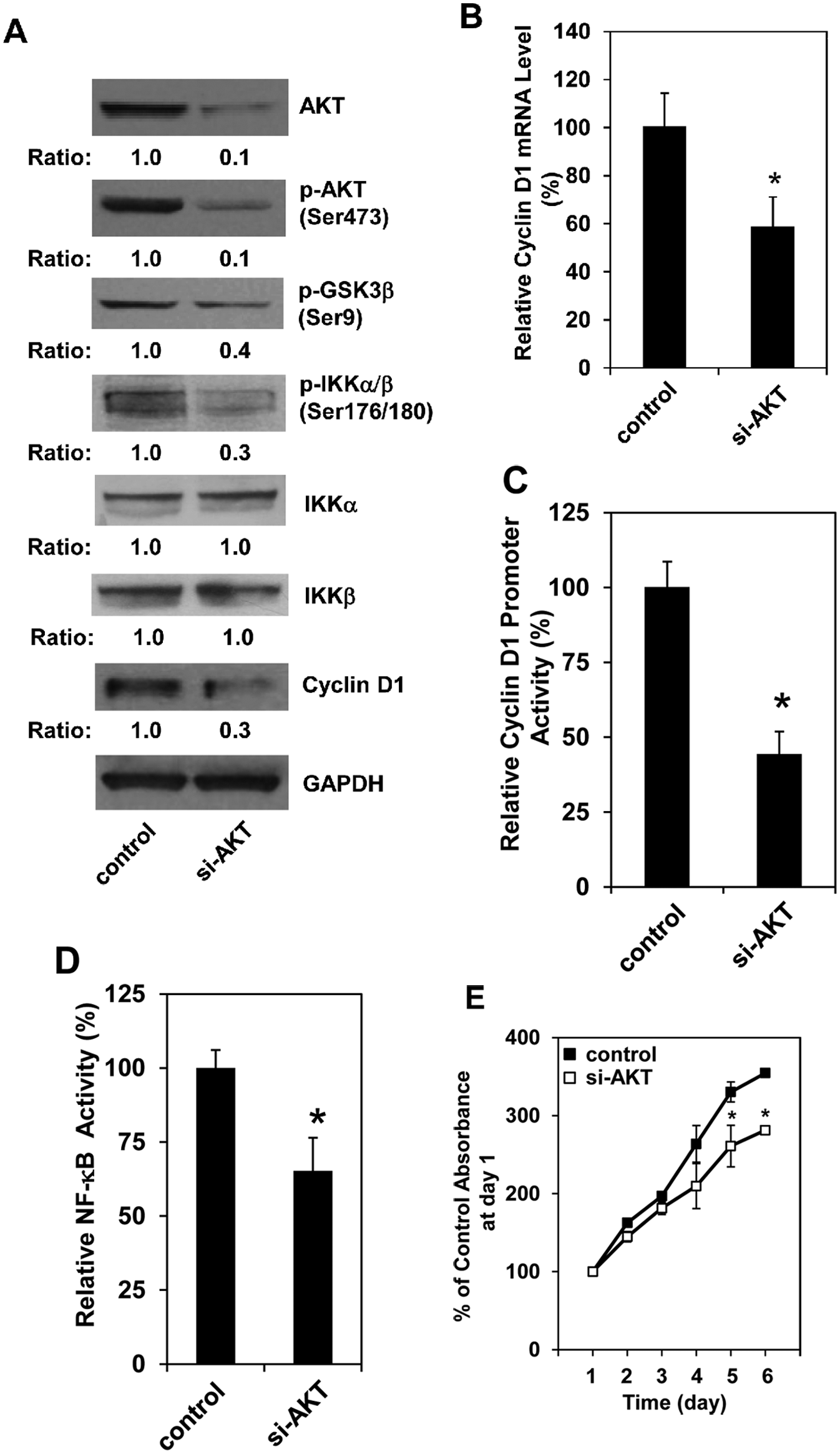
Knockdown of AKT inhibits cyclin D1 expression, NF-κB activation, and cell proliferation. Scramble siRNA (control) and akt siRNA (si-AKT) were transfected into HT29-shPdcd4 cells using transfection reagent according to the manufacturer’s protocol. (
Discussion
Induction of cyclin D1 and its binding to CDK4 and CDK6 is the rate-limiting step during cell cycle progression through G1 phase. The deregulation of this process is a key event in carcinogenesis and tumor progression. Elevation of cyclin D1 expression is frequently found in various types of human cancers, including breast, lung, and liver cancers. 31 In human colon cancer, cyclin D1 is elevated in about 30% of human adenocarcinomas and adenomatous polyps. 32 How does Pdcd4 regulate cyclin D1 expression? Our findings in this study suggest that the increased cyclin D1 expression in Pdcd4 knockdown cells is, at least in part, due to the NF-κB activation that is regulated by IKKα and IKKβ through AKT (Fig. 7). Furthermore, it has also been reported that cyclin D1 is a target gene of β-catenin–dependent transcription. 33 Thus, Pdcd4 might regulate cyclin D1 transcription via β-catenin–dependent transcription because Pdcd4 knockdown activates β- catenin–dependent transcription.7,8 In addition to transcriptional regulation, cyclin D1 expression can be regulated at the translational level. 34 Pdcd4 is a translation inhibitor that preferentially inhibits translation of mRNA with secondary structures at the 5′ untranslated region (5′UTR).35-37 The mRNAs for numerous growth factors and growth promotion genes, including cyclin D1, usually contain long GC-rich 5′UTR with the potential to form stable secondary structure(s) at the 5′ end. 38 Therefore, Pdcd4 might directly inhibit translation of cyclin D1. Pdcd4 is the binding partner of translation initiation factor (eIF) 4A.35,36 Inhibiting translation by Pdcd4 is believed to inhibit eIF4A’s helicase activity. 36 Recently, Wedeken et al. 39 showed that binding to eIF4A is also required for Pdcd4 associating with the 48S translation preinitiation complex. In addition, the phosphorylation of 4E binding protein (4EBP) and eIF4B, 2 AKT downstream targets, was increased in the Pdcd4 knockdown cells (unpublished data). Hyperphosphorylation of 4EBP dissociates the cap binding protein eIF4E and results in activation of cap-dependent translation. 40 Increased phosphorylation of eIF4B has also been shown to enhance translation and neoplastic transformation.41,42 Therefore, it is possible that Pdcd4 regulates cyclin D1 translation through activation of eIF4E and eIF4B (Fig.7). Whether Pdcd4 inhibits cyclin D1 translation needs further investigation.
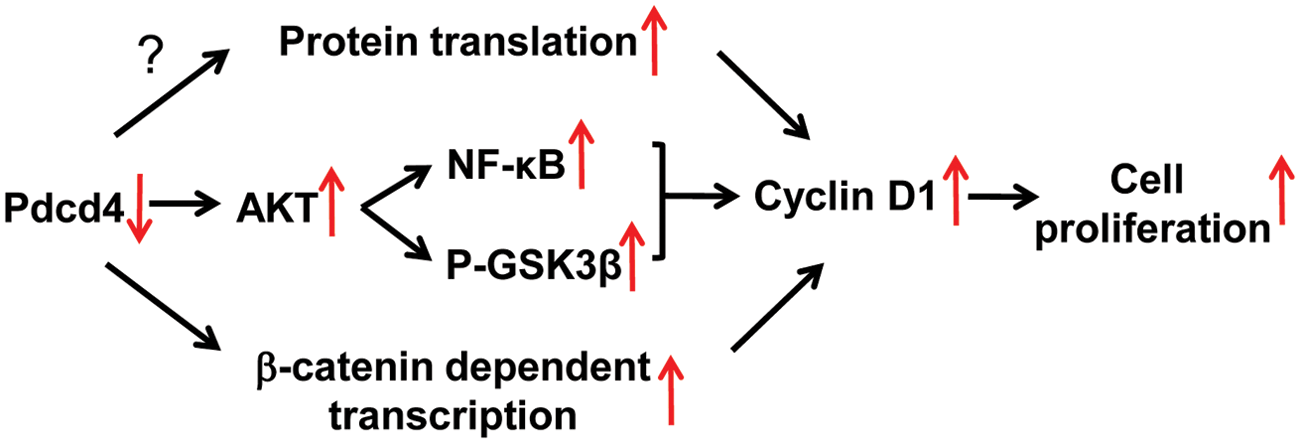
The scheme depicting the regulation pathways of cyclin D1 expression by Pdcd4 knockdown. See text for details.
AKT regulates a wide range of oncogenic processes, including cell growth and proliferation through phosphorylation of other protein kinases and transcription factors. AKT contains a central kinase domain with a threonine residue (Thr308) and a C-terminal regulatory site at serine 473 (Ser473). Phosphorylation of both Thr308 and Ser473 is essential for maximal AKT activation. 43 In cancer cells, AKT is constitutively activated. Immunohistochemical studies have shown that cancer cells expressing high levels of phospho-AKT (Ser473) were associated with poor prognosis in several types of cancer. 29 We report here that knockdown of Pdcd4 increases the level of phospho-AKT (Ser473) and overexpression of Pdcd4 inhibits the phosphorylation of AKT in colon tumor cell lines (Fig. 4), suggesting that Pdcd4 is able to regulate AKT activation. In addition, regulating AKT phosphorylation by Pdcd4 has also been observed in neuroendocrine Bon-1 cells. 44 Allgayer et al. 1 showed that the level of Pdcd4 and phospho-AKT (Ser473) is inversely correlated in human colon cancer tissues. These findings collectively suggest that the inhibitory role of Pdcd4 in human colon cancer development is at least partially achieved by inhibiting AKT activation. On the other hand, it has been reported that AKT is able to phosphorylate Pdcd4 at Ser67 and Ser457, and ribosomal protein S6 kinase, a downstream target of AKT, can phosphorylate Pdcd4 at Ser67.45,46 These findings reveal a feedback mechanism of regulation between Pdcd4 and AKT. Because AKT is a critical kinase during cancer development, agents targeted either upstream or downstream of the AKT signaling pathway for cancer therapy being actively developed. Our findings of Pdcd4 regulating AKT activation may provide a novel therapeutic approach for targeting AKT.
We also noted that knockdown of Pdcd4 up-regulates IKKα and IKKβ protein levels (Fig. 3B) and knockdown of AKT does not alter the expression of IKKα and IKKβ (Fig. 6A). These findings imply that Pdcd4 but not AKT regulates IKKα and IKKβ expression. In vitro studies have shown that the stem-loop structures with free energies (DG) of between –30 and –70 kcal/mol are sufficient to inhibit translation. 47 The DG values of the partial IKKα and IKKβ 5′ UTR are –39.7 kcal/mol and –47.3 kcal/mol (as calculated with the mfold program), respectively. Thus, it is possible that Pdcd4 regulates the translation of IKKα and IKKβ. It is also possible that Pdcd4 translationally regulates transcription factors that control IKKα and IKKβ expression. The mechanism of how Pdcd4 regulates IKKα and IKKβ expression needs further investigation.
Together, the current results demonstrate that knockdown of Pdcd4 promotes cell proliferation through AKT activation because knockdown of AKT in the Pdcd4 knockdown cells inhibits proliferation and cyclin D1 expression. Activation of AKT by Pdcd4 knockdown leads to up-regulation of cyclin D1 expression through NF-κB activation and GSK3b phosphorylation. These findings provide new insights into how loss of Pdcd4 expression promotes colon tumor development.
Materials and Methods
Cell culture
HT29-shLacZ and HT29-shPdcd4 cells were generated as described previously 7 and grown in McCoy’s medium containing 10% FBS, 2 mM L-glutamine, and 500 U/mL penicillin-streptomycin. LoVo-control and LoVo-Pdcd4 cells were generated as described previously 8 and grown in RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine, and 500 U/mL penicillin-streptomycin. Cells were incubated at 37°C with 5% CO2 in a humidified incubator.
Cell proliferation assay
Cell proliferation was measured using Cell Proliferation Kit II (XTT) (Roche Applied Science, Indianapolis, IN) as described previously. 42 Briefly, 3 × 103 cells/well were seeded onto 96-well plates. The sodium 3′-[1-(phenylaminocarbonyl)- 3,4-tetrazolium]-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate (XTT) labeling mixture was added to the cell culture for 2 hours. The concentration of water-soluble formazan salt was measured using the iMark microplate reader (Bio-Rad Laboratories, Hercules, CA) at 490 nm.
Cell cycle and annexin V apoptosis assay
For cell cycle assays, 1 × 106 cells were washed with PBS twice and then incubated with 70% (v/v) ethanol overnight. After washing with PBS, the cells were suspended in PBS containing 0.1% (v/v) Triton X-100, 40 mg/mL propidium iodide (Sigma, St. Louis, MO), and 50 mg/mL RNase A (Sigma) at 37°C for 30 minutes. Apoptosis assays were performed using the annexin V-FITC apoptosis kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Analysis was performed using a FacsCalibur cell analyzer (BD Biosciences, San Jose, CA).
Western blot analysis
Aliquots containing 20 to 40 mg of protein were separated onto SDS-PAGE and transferred to nitrocellulose membranes as described previously. 2 Subsequently, the membrane was incubated with primary antibodies overnight followed by horseradish peroxidase–linked secondary antibody for 1 hour. The target protein was visualized by chemiluminescence (Pierce, Rockford, IL). The antibodies against cyclin D1, phospho-AKT (Ser473), AKT (pan), phospho-GSK3β (Ser9), GSK3β, phospho-IKKα/β (Ser176/180), IKKα and IKKβ were purchased from Cell Signaling (1:1,000 dilution) (Danvers, MA). The GAPDH antibody was purchased from Santa Cruz Biotechnology (1:2,000 dilution) (Santa Cruz, CA). The phospho-AKT (Ser473) and AKT (pan) antibodies recognize all 3 AKT isoforms.
Real-time PCR
The total RNA isolation and real-time PCR were performed as described previously. 7 Briefly, after synthesis of the first-strand cDNA using the Superscript First-Strand Kit (Invitrogen), mRNA levels of cyclin D1 or GAPDH were quantified by real-time PCR in a LightCycler 480 (Roche Applied Science). The PCR cycling was performed at 95°C for 6 minutes followed by 40 cycles of denaturation (95°C for 15 seconds), annealing (61°C for 30 seconds), and extension (72°C for 20 seconds). To determine the specificity of the PCR, the amplified products were subjected to melt-curve analysis using the standard machine method. The target mRNA level was normalized to the internal control, GAPDH, using the formula ∆CT = CT (target) – CT (GAPDH) (CT = threshold cycle). The level of target gene expression in control cells was designated as 100%. The relative expression levels were calculated using the equation of 100 × 2–[average ∆CT (test) – average ∆CT (control)]. 48 The primers used for amplifying cyclin D1 and GAPDH were purchased from SA Biosciences (Frederick, MD).
Site-specific mutagenesis
The NF-κB binding sites on the cyclin D1 promoter were mutated using the wild-type cyclin D1 promoter fused with the luciferase construct (D1) as the template. The D1 construct was purchased from SwitchGear Genomics (Menlo Park, CA), which includes –920 bp to +106 bp of cyclin D1 promoter region (relative to the transcription start site) fused with the luciferase reporter. The site-specific mutagenesis was performed using QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions.
AKT kinase activity assay
AKT kinase assays were performed using the AKT kinase assay kit (Cell Signaling) according to the manufacturer’s protocol. Briefly, cells were lysed in 500 mL lysis buffer and incubated on ice for 1 hour. After centrifugation, an aliquot (500 mg) of supernatant was added to 20 mL of immobilized AKT Ser473 antibody beads and incubated with gentle rocking overnight at 4°C. The beads were then washed with 500 mL of 1x lysis buffer twice and 500 mL of 1x kinase assay buffer twice. The beads were suspended in 50 mL of 1x kinase assay buffer containing 200 mM of ATP and 2 mg of recombinant GSK-3 fusion protein and incubated for 30 minutes at 30°C. The reaction was terminated by adding 25 mL of 4x sodium dodecyl sulfate sample buffer. The proteins were separated onto a 4% to 12% Bis-Tris polyacrylamide gel (Invitrogen) and immunoblotted using phospho-GSK3α/β (Ser21/9) antibody. The band intensity was quantified using VisionWork LS image acquisition and analysis software (UVP, Upland, CA).
Transient transfection and luciferase activity assays
Cells (3 × 104 cells/well in 24-well plates) were transiently transfected with 0.2 mg of luciferase reporter plasmid along with 10 ng of pRL-SV40 (Promega, Madison, WI) by using TurboFect in vitro transfection reagent (Fermentas, Glen Burnie, MD). After 48 hours, the cells were lysed, and the luciferase activity was determined as described previously. 7 The luciferase activity of each sample is normalized against Renilla luciferase activity for monitoring transfection efficiency.
Knockdown of IKK and AKT
HT29-shPdcd4 cells (2 × 105 cells on a 60-mm plate) were transfected with 22 mL of 10 mM IKKα siRNA (Santa Cruz Biotechnology), IKKβ siRNA (Santa Cruz Biotechnology), akt siRNA (Santa Cruz Biotechnology), or scramble siRNA (negative control) (Invitrogen) using INTERFERin transfection reagent (Polyplus-Transfection Inc., New York, NY) according to the manufacturer’s protocol. For Western blot assays, cells were collected at day 4 after transfection. For transfection assays, cells were split at day 2 after transfection.
Footnotes
Acknowledgements
The authors gratefully thank Yan Zhang for the technical assistance.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported by the National Institutes of Health [grant number R01CA129015].