
Abstract
SIRT3 is a NAD+-dependent deacetylase that regulates the function of numerous mitochondrial proteins with roles in metabolism, oxidative stress, and cell survival. It is emerging as an instrumental regulator of the mitochondrial adaptive responses to stress, including metabolic reprogramming and enhancing antioxidant defense mechanisms. Here, we discuss the role that SIRT3 plays at both a cellular and physiological level and consider its involvement in disease. Mitochondrial dysfunction is a key contributing factor in many diseases; however, the mechanisms involved are often not well understood, and few targeted therapies exist. If manipulation of SIRT3 proves to be beneficial in disease states, then it could be a promising target for novel therapies.
Introduction
An aging population, coupled with increasing obesity rates, has presented a number of new challenges to society, particularly due to the increased incidence of diseases for which age and obesity are major risk factors. This has placed a huge burden upon the health care systems of developed countries in particular, and there is a dire need for more effective therapies. Mitochondrial dysfunction is a common feature of many of these diseases, including cancer, neurodegenerative diseases, and metabolic disorders; thus, fully understanding the roles that mitochondria play in these diseases should be a priority for the development of novel therapies. Identification of the causes of mitochondrial dysfunction, as well as of cell-protective mechanisms that respond to the dysfunction, will be pivotal for finding strategies to both treat and delay the onset of mitochondrial-mediated diseases. As central regulators of metabolic function and cell survival, the sirtuins are emerging as promising targets to lead these investigations.
The sirtuins are a highly conserved family of proteins, most of which possess NAD+-dependent protein deacetylase activity. 1 The yeast ortholog SIR2 was originally reported to regulate the life span, a finding that spurred a high volume of research into sirtuins. 2 Although the significance of their role in the regulation of the life span has been challenged recently, 3 it has become clear that the sirtuins are involved in a number of diseases, including age-related diseases, and that modulation of their function may have beneficial effects. The sirtuins are also thought to mediate some of the protective effects of calorie restriction (CR), a dietary regimen widely documented to extend the life span as well as improve the health span by delaying the onset of age-related diseases. 1 It may therefore be possible to harness some of the benefits of CR via the modulation of sirtuin function.
Of the 7 mammalian sirtuins, SIRT3, SIRT4, and SIRT5 localize to the mitochondria. This means that they are ideally situated for targeted therapeutic intervention, heightening the importance of appreciating the regulation and function of this subset of sirtuins. SIRT3 is thought to be the primary mitochondrial deacetylase as, in contrast to SIRT4- and SIRT5-deficient mice, SIRT3-deficient mice display hyperacetylation of mitochondrial proteins in the liver and brown adipose tissue. 4 A recent study employing mass spectrometry revealed that as many as 65% of mitochondrial proteins are lysine acetylated, suggesting an important regulatory role for the modification. 5 In addition, the profile of acetylated proteins was markedly different in SIRT3-deficient mice compared to wild-type (wt) mice. 5 SIRT3 has now been shown to regulate many aspects of mitochondrial function, including metabolism, ATP generation, and modulation of the response to oxidative stress. Together, these properties place SIRT3 at the fulcrum of mitochondrial regulation. In this review, we will discuss the various roles played by SIRT3 via the regulation of its substrates at the mitochondrial level and extend this to the broader physiological implications of SIRT3 function. We will also speculate on the therapeutic potential for manipulating SIRT3 in the treatment of disease.
SIRT3-Dependent Regulation of Mitochondrial Metabolism and Energy Homeostasis
The first substrates identified for SIRT3, acetyl-CoA synthetase 2 (AceCS2) and glutamate dehydrogenase (GDH), indicated that SIRT3 has a regulatory role in mitochondrial metabolism.4,6 AceCS2 localizes to the mitochondria and is activated by deacetylation, whereupon it converts acetate to acetyl-CoA, which can then be used in metabolic pathways including the citric acid cycle. 6 SIRT3 deacetylates AceCS2 both in vitro and in vivo, resulting in increased AceCS2 activity.7,8 AceCS2 may be of particular importance during starvation or times of low food availability as it allows free acetate to be used in place of pyruvate produced by glycolysis to generate acetyl-CoA, which is required to fuel the citric acid cycle. 9 SIRT3-dependent deacetylation also increases the enzymatic activity of GDH in vitro and in vivo.4,9 GDH is involved in the oxidation of amino acids, allowing them to be used as fuel in the citric acid cycle. Interestingly, a shift away from dependence on liver glycolysis, facilitated by both GDH and AceCS2 activity, has been implicated in CR. 9 This is suggestive of a role for SIRT3 in reprogramming metabolism during CR to allow respiration to continue efficiently during times of low food availability. The subsequent identification of 3-hydroxy-3-methylglutaryl CoA synthase, long-chain acyl-CoA dehydrogenase, and ornithine transcarbamylase as SIRT3 substrates suggests further regulatory roles for SIRT3 in ketogenesis, fatty acid oxidation, and the urea cycle.10-12 Furthermore, a study by Hirschey et al. 13 reported that a single nucleotide polymorphism in the human Sirt3 gene is associated with increased susceptibility to the development of the metabolic syndrome, a condition to which SIRT3-deficient mice are also predisposed. Collectively, these findings place SIRT3 as a central regulator of mitochondrial metabolism, particularly in controlling energy substrate utilization in response to altered nutrient availability.
SIRT3 further modulates energy homeostasis in the mitochondria by regulating ATP generation from oxidative phosphorylation. Mechanistically, SIRT3 has been shown to interact with subunits of complex I and II of the electron transport chain (ETC), resulting in increased activity of both complexes.14,15 A proteomics-led study also revealed 2 subunits of ATP synthase as likely interacting partners of SIRT3, which is confirmed in HEK293 cells where SIRT3 overexpression reduced acetylation levels of ATP synthase. 16 Furthermore, mouse embryonic fibroblasts (MEFs) and a number of organs from SIRT3−/− mice were shown to have decreased basal ATP levels. 14
The mitochondrial ribosomal protein MRPL10 was recently identified as a SIRT3 target, suggesting a wider regulatory role for SIRT3 in mitochondrial respiration. 17 The mitochondrial ribosomal proteins are required for the synthesis of proteins encoded by mitochondrial DNA, which includes 13 components of the ETC. As well as deacetylating MRPL10, SIRT3 was also shown to interact with the 55S mitochondrial ribosome. Interestingly, deacetylation of MRPL10 by SIRT3 was associated with decreased mitochondrial ribosome activity: ribosomes isolated from SIRT3−/− mice possessed increased translation activity, whereas SIRT3 overexpression in a cell line decreased mitochondrial protein synthesis. 17 Together, these data suggest a complex role for SIRT3 in the regulation of activity and expression of ETC components and demonstrate the need for additional mechanistic studies of SIRT3 function in vivo.
Despite regulating the activity of numerous mitochondrial proteins, SIRT3-deficient mice appear to be physiologically unaffected under basal conditions, with no differences in body weight or fat content compared to wt littermates.4,14 This is somewhat surprising given that altered mitochondrial function, including impaired fatty acid oxidation and defects in the ETC, can lead to hepatic steatosis in both humans and mice as well as alterations in body composition. 4 However, SIRT3-deficient mice fare worse than wt mice in response to metabolic stresses, displaying cold intolerance following a period of fasting and developing more severe hepatic steatosis when fed a high-fat diet.11,13 This is consistent with a requirement for SIRT3 in reprogramming metabolism and mounting adaptive responses to stress, discussed in more detail below.
SIRT3-Dependent Regulation of Oxidative Stress and Cell Survival
SIRT3 is central in the maintenance of appropriate mitochondrial function not only by regulating energy homeostasis but also, as recent studies have shown, by limiting oxidative stress. This is crucial for the protection of cellular components from oxidative damage that can contribute to diminished function and ultimately cell death.
Shi et al. 18 first reported that overexpression of SIRT3 reduced reactive oxygen species (ROS) production in an adipocyte cell line, which was accompanied by a decrease in mitochondrial membrane potential. This observation was supported by a study that identified a role for SIRT3 in cardiac hypertrophy; SIRT3 overexpression in cardiomyocytes resulted in decreased mitochondrial ROS production, while cardiomyocytes from SIRT3−/− mice had increased ROS production. 19 The expression of 2 antioxidant proteins, mitochondrial superoxide dismutase (SOD2) and catalase, was increased in mice overexpressing SIRT3. 19 SOD2 catalyzes the first step of superoxide detoxification by converting superoxide to H2O2, while catalase converts H2O2 to water. 19 FOXO3a, a transcription factor that upregulates the expression of SOD2 and catalase, was deacetylated by SIRT3 in cardiomyocytes. Deacetylation is thought to promote nuclear localization of FOXO3a and thus provides a mechanism by which SIRT3 may decrease ROS levels. 19 FOXO3a shuttles between the cytoplasm and nucleus; this raises interesting questions concerning the spatial coordination of FOXO3a regulation by SIRT3, as the form of SIRT3 overexpressed in cardiomyocytes and demonstrated to interact with FOXO3a localizes exclusively to the mitochondria.19,20 Interestingly, a second study found FOXO3a to be present in mitochondrial fractions; however, SIRT3-mediated deacetylation of FOXO3a at the level of the mitochondria remains to be demonstrated in vivo. 21
Two direct mechanisms linking SIRT3 with reduced ROS production have been proposed following the identification of isocitrate dehydrogenase 2 (IDH2) and SOD2 as direct targets for SIRT3.22-24 IDH2 is deacetylated by SIRT3 in vitro, leading to an increase in IDH2 activity. 9 A subsequent study by Someya et al. 22 found that while CR decreased IDH2 acetylation levels in the liver of wt mice, this effect was absent in SIRT3−/− mice. Co-immunoprecipitation of exogenously expressed SIRT3 and IDH2 together in HEK293 cells further suggests that SIRT3 directly deacetylates IDH2. 22 IDH2 catalyzes the conversion of isocitrate to α-ketoglutarate in the mitochondria, generating nicotinamide adenine dinucleotide phosphate (NADPH) from the oxidized form NADP+ in the process. NADPH is required for the regeneration of reduced glutathione, another of the cell’s main antioxidants. Therefore, increased IDH2 activity enhances the glutathione antioxidant defense system. 25 SIRT3 also deacetylates SOD2, resulting in increased SOD2 activity.23,24 Furthermore, MEFs and liver tissue from SIRT3−/− mice had reduced SOD2 activity and increased ROS production, while SIRT3 overexpression reduced ROS production in wt but not in SOD2−/− MEFs.23,24 The diminished defenses of SIRT3-deficient mice against oxidative stress have thus far been shown to contribute to genomic instability, age-related neuronal loss, and a tumor- permissive environment, highlighting the importance of SIRT3 in cell-protective pathways.22,23,26
A number of studies have pointed to a role for SIRT3 in cell survival. In addition to limiting ROS levels, this may also be due to SIRT3 regulating the formation and opening of the mitochondrial permeability transition pore (mPTP) via deacetylation of cyclophilin D (cypD), one of the key components of the pore. 27 The mPTP is a transmembrane channel comprising several proteins including the adenine nucleotide translocator (ANT) that assemble at contact sites between the inner and outer mitochondrial membranes. Its formation leads to a rapid loss of mitochondrial membrane potential, mitochondrial swelling, release of cytochrome c and other small proteins, and ultimately cell death. 27 Deacetylation of cypD by SIRT3 is thought to reduce cypD binding to ANT, thereby restraining mPTP formation. 27 In addition, SIRT3-catalyzed deacetylation of cypD promotes the detachment of hexokinase II, a mediator of the mPTP, from the mitochondria. 28 When bound to the mitochondria, hexokinase II leads to an increased rate of glycolysis while decreasing oxidative phosphorylation. These are common features of many cancer cells, suggesting a further cell-protective role for SIRT3 in the prevention of cancer development. 28
Collectively, these findings suggest that SIRT3 is an important regulator of the mitochondrial responses to oxidative stress and mitochondrial-mediated cell death. In line with this, a number of neuroprotective roles have been identified for SIRT3. Overexpression of SIRT3 in primary cortical cultures protected neurons from NMDA-mediated excitotoxicity, 29 and in a cellular model of amyotrophic lateral sclerosis, SIRT3 overexpression reduced mitochondrial fragmentation and cell death. 30 In addition, primary hippocampal neurons overexpressing SIRT3 were protected against death induced by inhibition of the ETC, shown to lead to an increase in mitochondrial ROS production. 31 These studies highlight an intriguing role for SIRT3 in the central nervous system, which could have implications for a range of disorders in which neuronal health is compromised. Further studies will be required to determine if SIRT3 is neuroprotective in vivo and address whether overexpression of SIRT3 could be a strategy for maintaining neuronal health.
SIRT3 in Disease
Consistent with its role in regulating mitochondrial function and in particular oxidative stress, SIRT3 has been implicated in disease states including cancer and neurodegenerative disease.26,30-33
Cancer is associated with the metabolic reprogramming of cells, orchestrated by various mechanisms including transcriptional changes induced by the transcription factor hypoxia inducible factor-1α (HIF-1α). Ordinarily targeted for degradation under normoxic conditions, HIF-1α is stabilized when levels of ROS are increased. Upon stabilization, HIF-1α translocates to the nucleus and induces the transcription of its target genes, including several that are linked with various aspects of tumorigenesis. 34 SIRT3 acts as a tumor suppressor by limiting ROS levels, thereby leading to the destabilization and subsequent degradation of HIF-1α.32,33 In addition, SIRT3−/− MEFs have increased chromosomal instability due to increased superoxide levels. 26 Collectively, these studies suggest that a loss of SIRT3 contributes to a tumor-permissive environment. In support of this, SIRT3-deficient mice spontaneously develop mammary gland tumors with age, and SIRT3 expression is reduced in human breast cancers.26,33
Increased oxidative stress and mitochondrial dysfunction are also prominent features of many neurodegenerative disorders, including Alzheimer disease (AD). 35 It was recently found that SIRT3 expression is increased in both human and mouse AD pathology. 31 SIRT3 expression was also upregulated in response to increased ROS production in primary neuronal cultures, suggesting that SIRT3 may be upregulated as part of the mitochondrial responses to increased oxidative stress in AD. 31 Oxidative stress also contributes to age-related hearing loss, one of the phenotypes associated with aging. CR prevents age-related hearing loss in mice by protecting cochlear neurons from oxidative damage and death and is completely dependent on SIRT3 to do so. 22 Given that SIRT3 expression is increased by CR, this further suggests that SIRT3 is upregulated as part of a cell-protective mechanism in response to oxidative stress.22,36
The aging process itself is associated with increased oxidative stress and mitochondrial dysfunction. SIRT3 is unique among the sirtuins in that it is the only one to have been directly linked to the regulation of the human life span. A study of variability within the Sirt3 gene identified a variable number tandem repeat (VNTR) polymorphism with associated allele-specific enhancer activity. 37 Analysis of correlation between age and allele frequency revealed that male individuals over 90 years of age had almost no alleles lacking VNTR-associated enhancer activity, suggesting a correlation between SIRT3 expression and an enhanced life span. 37 The association between SIRT3 and the life span has not been investigated in mouse studies. Indeed, the focus of sirtuin research has to some extent shifted away from regulation of the life span and towards the role of sirtuins in disease. However, a recent study by Kanfi et al. 38 reporting on regulation of the life span in mice by SIRT6 may well serve to reinvigorate this arm of research in the sirtuin field. With the development of models overexpressing SIRT3, it will be of interest to learn whether the life span is altered in these mice in addition to the impact that SIRT3 overexpression may have in disease. These may of course be intricately linked, given that aging is a major risk factor for numerous age-related diseases.
SIRT3: Dispensable until Times of Stress?
Perhaps one of the most notable features of SIRT3 is that it appears to be of primary importance in the adaptive responses to various forms of environmental challenge or stress. A number of studies performed in SIRT3−/− mice have shown that these mice appear indistinguishable from wt mice under basal conditions. While some differences have been reported between SIRT3−/− and wt mice in the basal state at a biochemical level—mitochondrial proteins were hyperacetylated, and basal ATP levels were reduced in a number of tissues from SIRT3−/− mice4,13,14—this does not appear to be translated into an overt physiological phenotype. The importance of SIRT3 is apparent, however, when examining the responses to age- and diet-related changes. Someya et al. 22 examined a range of markers of oxidative damage and antioxidant capacity and found no significant differences between wt and SIRT3−/− mice on a control diet. However, unlike wt mice fed a calorie-restricted diet, calorie-restricted SIRT3−/− mice were not protected from age-related oxidative damage and showed no improvement in antioxidant capacity. 22 A different study using SIRT3−/− mice examining the effects of a high-fat diet found that SIRT3−/− mice developed diet-induced obesity at an accelerated rate compared to wt littermates as well as being insulin resistant on both a standard and high-fat diet. 13 Interestingly, however, mice with liver- or muscle-specific SIRT3 deficiency remained indistinguishable from wt mice even after being fed a high-fat diet; therefore, further studies will be required to resolve the tissue-specific actions of SIRT3. 39
The requirement for SIRT3 for cellular protection under stress conditions is further supported by studies that have investigated the regulation of SIRT3 expression. CR, fasting, exercise, and genotoxic and oxidative stress all increase SIRT3 expression.11,18,29,31,40,41 Collectively, these data suggest that SIRT3 is upregulated as part of a stress-responsive, cell-protective mechanism. SIRT3-mediated deacetylation results in increased activity of the majority of its target proteins. The data from SIRT3-deficient mice suggest that under basal conditions, target proteins in their acetylated form possess sufficient activity. However, under conditions that lead to challenges such as increased oxidative stress, a lack of SIRT3 deacetylation may mean that the activity of SIRT3 target enzymes required for protection (including increased antioxidant capacity and mitochondrial respiration) cannot be increased sufficiently to meet the demand, as summarized in Figure 1. Therefore, identification of the crucial substrates for SIRT3 in the context of particular stresses will be needed to understand the pathways mediating cell protection.
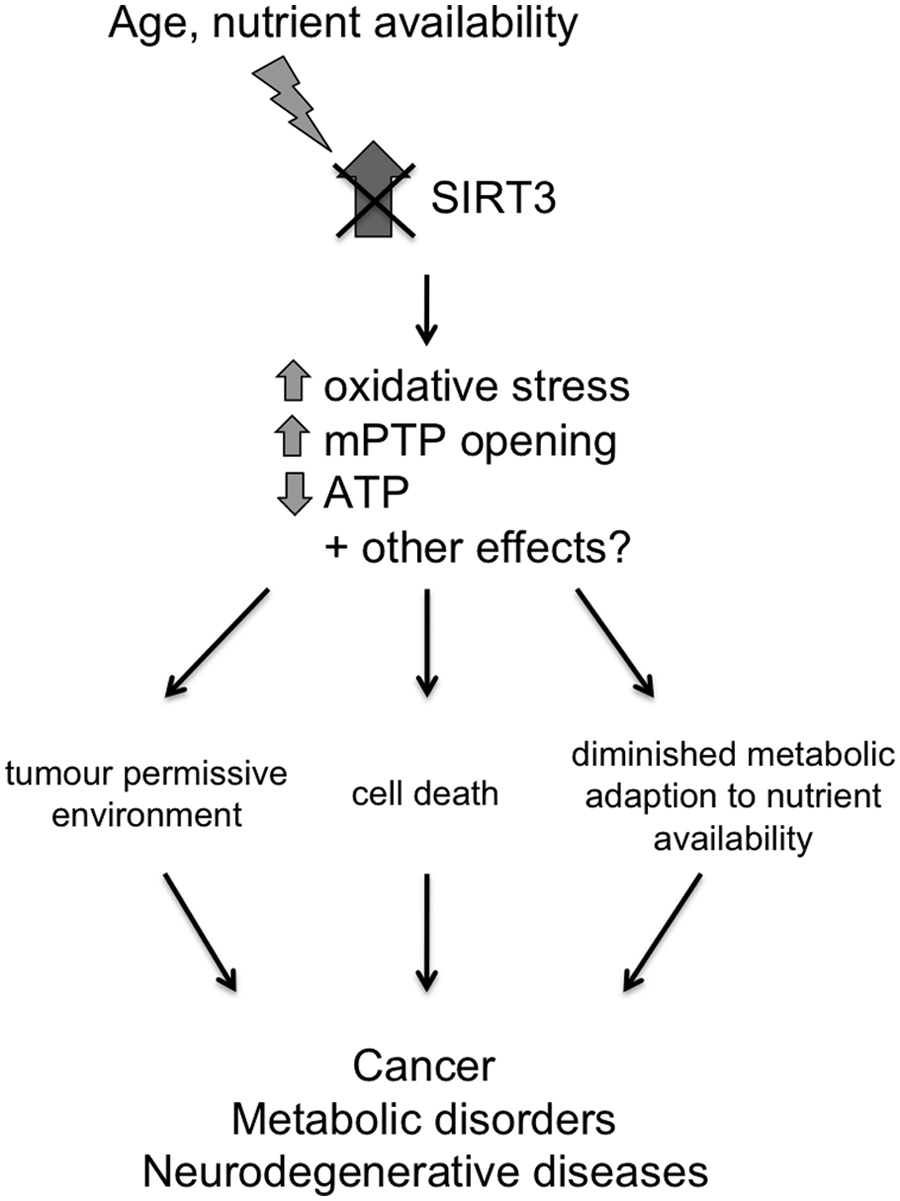
SIRT3 is required to mount adaptive mitochondrial responses to stress. SIRT3-deficient mice are less able to mount adaptive responses to external stresses such as aging and altered nutrient availability. Hyperacetylation of SIRT3 substrates results in increased oxidative stress and opening of the mPTP and decreased ATP levels, leading to genomic instability and cell death. A failure to trigger cell-protective mechanisms to counteract the stress may contribute to the development of cancer, neurodegenerative diseases, and metabolic disorders.
The effects of mitochondrial SIRT3 overexpression in vivo are yet to be reported. Undoubtedly, such studies will reveal whether, as predicted, SIRT3 upregulation occurs as part of a cell-protective response. They will also be invaluable in determining if increasing SIRT3 expression or activity could be a beneficial intervention in the treatment of diseases associated with mitochondrial dysfunction.
Molecular Mechanisms Controlling SIRT3 Expression
As described above, the physiological conditions that lead to an alteration in SIRT3 expression are emerging. However, the molecular mechanisms underpinning this are less clear. Some insight into this was provided by the finding that peroxisome proliferator–activated receptor γ co-activator-1α (PGC-1α), a transcription factor that regulates mitochondrial biogenesis, also regulates SIRT3 expression via estrogen-related receptor-α (ERRα) 42 ; a putative ERRα binding element was identified in the promoter region of SIRT3, and ERRα binding mediates the transcription of SIRT3 induced by PGC-1α in C2C12 cells and mouse hepatocytes. 42 This was supported by Hirschey et al., 13 who showed that, as well as leading to reduced SIRT3 expression, chronic high-fat diet feeding resulted in reduced hepatic PGC-1α expression in mice. Furthermore, overexpression of PGC-1α increased SIRT3 expression in high-fat diet–fed mice. Interestingly, PGC-1α overexpression had no effect on SIRT3 expression in mice fed a standard diet, suggesting that additional factors or triggers are involved for SIRT3 regulation by PGC-1α. 13 SIRT1 regulates PGC-1α activity by direct deacetylation, resulting in an increase in PGC-1α transcriptional activity both in vitro and in vivo.43,44 Given that SIRT3 appears to be under the transcriptional control of PGC-1α under certain conditions, this may point towards a mechanism by which SIRT1 regulates the expression of SIRT3, albeit indirectly.
SIRT3 is specifically upregulated in response to increased ROS production. 31 This upregulation could conceivably be controlled by an oxygen-sensing mechanism involving a transcription factor such as HIF-1α. As discussed above, SIRT3 mediates HIF-1α stabilization via the regulation of ROS levels.26,32,33 If SIRT3 is indeed a target gene of HIF-1α, then it would be tempting to speculate that a negative feedback mechanism may exist whereby HIF-1α upregulates SIRT3, which in turn decreases ROS levels, resulting in degradation of HIF-1α.
Another pathway that regulates the transcription of a number of cell-protective genes in response to oxidative stress is the nuclear factor E2-related factor 2 (Nrf2) antioxidant response element signaling pathway. Upon oxidative stress, Nrf2 translocates to the nucleus and induces the expression of genes that have an antioxidant response element binding site. 45 Interestingly, like SIRT3, Nrf2 has been implicated in regulating energy metabolism and mitochondrial biogenesis. 46 It is clear that further studies will be required to determine what mechanisms are responsible for altering SIRT3 expression, and analysis of the DNA upstream of SIRT3 in its promoter region may help in establishing this. Ultimately, targeting these mechanisms may be a potential route to manipulating SIRT3 levels if this proves to be beneficial for therapeutic intervention in disease states.
Modulation of SIRT3 Activity
A different approach to harness the beneficial effects of SIRT3 is to increase its activity rather than its expression level. In the current absence of any direct activators of SIRT3, it may be possible to achieve this by increasing mitochondrial NAD+ levels. This was shown to be the case in a recent study by Canto et al., 47 who found that feeding mice the NAD+ precursor nicotinamide riboside (NR) led to increased NAD+ levels, including an increase in the mitochondrial pool of NAD+. Moreover, NR treatment led to a concomitant increase in SIRT3 activity. 47 However, these effects were restricted to certain tissues; while NAD+ levels and SIRT3 activity were increased in the liver, no differences were found in the brains of NR-fed mice compared to controls. 47 Furthermore, SIRT1 activity was also activated by NR feeding due to increased nuclear NAD+ levels, and so this approach does not enable specific activation of SIRT3. At the very least, this study shows that SIRT3 activity can be increased in vivo by increasing mitochondrial NAD+ levels and is a promising step forward in the field of sirtuin activation. Future studies will be required to determine if it is possible to specifically activate SIRT3 by increasing NAD+ availability only in the mitochondria. In addition, to enable the manipulation of SIRT3 activity in the brain, it will be necessary to develop NAD+ precursors or small molecule activators to act there, which will be dependent on their ability to cross the blood-brain barrier.
As studies begin to focus on manipulating SIRT3 function, it will be important to establish whether altering SIRT3 expression or activity alone is sufficient or whether a combined approach may be necessary in some circumstances. This may well be dependent on factors such as basal SIRT3 expression and NAD+ levels, which vary between tissues, disease states, and in response to energy availability.18,31,48 Figure 2 summarizes some potential strategies for modulating SIRT3 expression and activity.
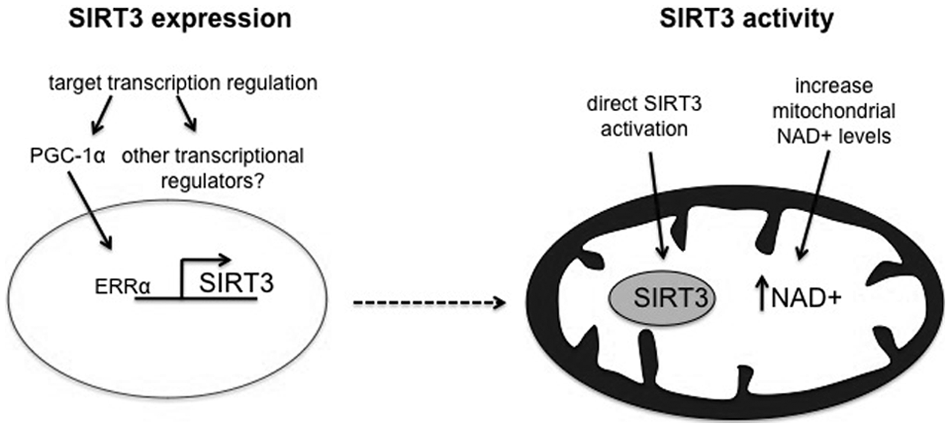
Speculative strategies to harness the cell-protective effects of SIRT3. If modulation of SIRT3 function proves beneficial, manipulating its expression and function could be targets for therapeutic intervention. It may be possible to increase SIRT3 expression by targeting the PGC-1α/ERRα axis or other as yet unidentified transcriptional regulators of SIRT3. Enhancing SIRT3 activity could be achieved via direct activation or by increasing mitochondrial NAD+ levels.
Summary
While our understanding of SIRT3 function is undoubtedly incomplete, great strides forward have been made in recent years by the identification of numerous SIRT3 targets and by studies investigating the implications of loss of SIRT3. This paves the way for future studies to determine whether modulation of SIRT3 function may work as a cell-protective mechanism via enhancing mitochondrial function. The prevalence of mitochondrial dysfunction in disease coupled with the instrumental role that SIRT3 appears to play in allowing adaptive responses to be mounted suggest that SIRT3 may be a pivotal point of intervention in the development of novel therapeutics.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: H.J.M.W. was funded by a BBSRC CASE (Biotechnology and Biological Sciences Research Council Collaborative Awards in Science and Engineering) PhD studentship, with support from Eli Lilly & Co. Ltd. to N.B. and J.D.L. N.B. was funded by a Lister Institute for Preventive Medicine Prize Fellowship.