
Abstract
SIRT1 is a NAD+-dependent protein deacetylase that has a very large number of established protein substrates and an equally impressive list of biological functions thought to be regulated by its activity. Perhaps as notable is the remarkable number of points of conflict concerning the role of SIRT1 in biological processes. For example, evidence exists suggesting that SIRT1 is a tumor suppressor, is an oncogene, or has no effect on oncogenesis. Similarly, SIRT1 is variably reported to induce, inhibit, or have no effect on autophagy. We believe that the resolution of many conflicting results is possible by considering recent reports indicating that SIRT1 is an important hub interacting with a complex network of proteins that collectively regulate a wide variety of biological processes including cancer and autophagy. A number of the interacting proteins are themselves hubs that, like SIRT1, utilize intrinsically disordered regions for their promiscuous interactions. Many studies investigating SIRT1 function have been carried out on cell lines carrying undetermined numbers of alterations to the proteins comprising the SIRT1 network or on inbred mouse strains carrying fixed mutations affecting some of these proteins. Thus, the effects of modulating SIRT1 amount and/or activity are importantly determined by the genetic background of the cell (or the inbred strain of mice), and the effects attributed to SIRT1 are synthetic with the background of mutations and epigenetic differences between cells and organisms. Work on mice carrying alterations to the Sirt1 gene suggests that the network in which SIRT1 functions plays an important role in mediating physiological adaptation to various sources of chronic stress such as calorie restriction and calorie overload. Whether the catalytic activity of SIRT1 and the nuclear concentration of the co-factor, NAD+, are responsible for modulating this activity remains to be determined. However, the effect of modulating SIRT1 activity must be interpreted in the context of the cell or tissue under investigation. Indeed, for SIRT1, we argue that context is everything.
The risk of many important diseases increases dramatically with age. Calorie restriction (CR) of experimental animals prolongs the life span and forestalls the onset of a remarkably diverse spectrum of diseases such as cancer, cardiovascular disease, neurodegenerative conditions, autoimmunity, and metabolic diseases including diabetes. 1 Work on small eukaryotic organisms has established that the beneficial effects of CR depend on a class of enzymes called sirtuins. The working model is that CR activates sirtuin function. In mammals, SIRT1 is the most studied sirtuin. SIRT1 activity in human tissues normally declines with age, 2 consistent with the idea that SIRT1 has a role in the age-dependent increase in disease susceptibility and with the notion that increasing SIRT1 activity might prolong the health span and resistance to age-dependent disease.
This article will attempt to resolve some of the many inconsistencies in the literature regarding the roles of SIRT1. It is our conviction that the intact organism is the appropriate one in which to investigate the physiological roles of SIRT1 and that genetic alteration to the endogenous Sirt1 gene constitutes the cleanest form of experimental manipulation. The reader is referred to a recent review of the sirtuin field 3 for an excellent encyclopedic summary of the literature.
The Biochemistry of Sirtuins
The sirtuin family of proteins is characterized by the presence of a catalytic domain that couples NAD+ hydrolysis to protein deacetylation.4-6 This domain is found in genes of all animal and plant phyla.7-9 The yeast sir2 protein is the founding member of this family and is known to deacetylate histones H4 (K16Ac) and H3 (K9Ac) to facilitate the establishment of transcriptionally inactive chromatin.10,11 In addition, sir2 confers an enhanced resistance to cellular stresses and an increased replicative life span. 12 The biochemical pathways responsible for these characteristics may be related to sir2-mediated inhibition of recombination between members of the ribosomal gene cluster13,14 or may be independent of chromatin effects. 15 The sir2 orthologous genes in Caenorhabditis elegans and Drosophila melanogaster have also been reported to prolong the life span and increase organismal resistance to stress,16,17 although these observations, like many in the sirtuin field, are controversial. 18
Of the 7 sirtuins encoded in the mammalian genome, the SIRT1 protein has the amino acid sequence most closely related to sir2 and is thought to be its ortholog. SIRT1 is a nuclear protein expressed in all tissues, albeit at variable levels, 19 and it has been the object of intense study. Unlike other members of the sirtuin family, the core sirtuin domain of SIRT1 is insufficient for catalytic activity. Another 25–amino acid peptide called essential for SIRT1 activity (ESA) is also required for catalysis. 20
The spectrum of acetylated protein substrates of SIRT1 is vast; at least 79 have been described (Table 1) including a large number of important nuclear proteins (such as p53, Rb, NF-κB, and CBP/p300) along with a variety of cytoplasmic proteins. The identification of cytoplasmic substrates for a protein that appears to be primarily nuclear in interphase cells is one of the many apparent paradoxes in the field. The SIRT1 enzyme is able to deacetylate acetyllysines in peptides with little context-dependent substrate specificity 21 in vitro, so establishing what is and what is not a SIRT1 substrate can be problematic.
The Gene Products That Have Been Established as Acetylated Proteins Capable of Being Deacetylated by SIRT1 Either In Vivo, In Vitro, or Both
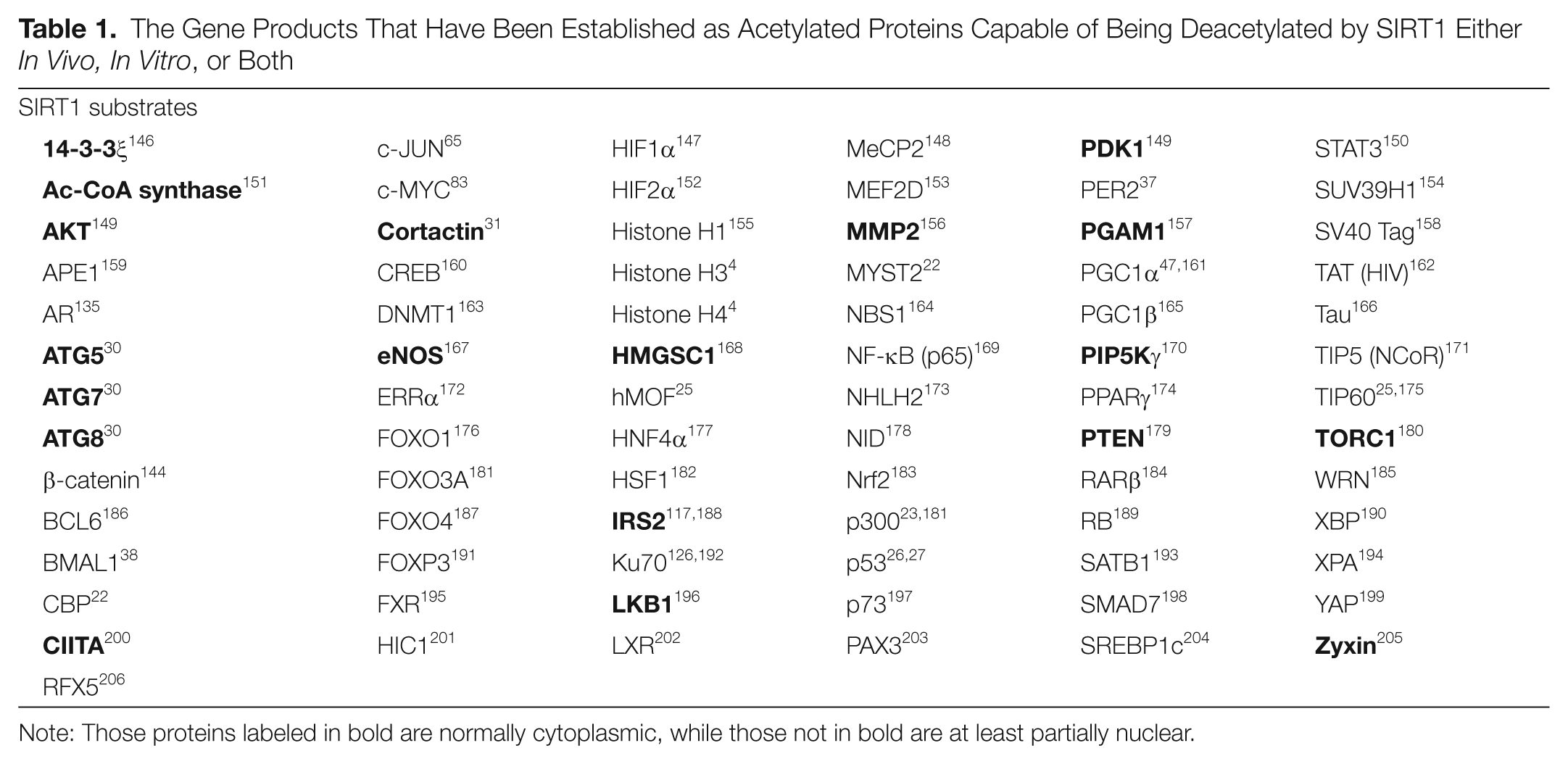
Note: Those proteins labeled in bold are normally cytoplasmic, while those not in bold are at least partially nuclear.
A recent investigation of the “acetylome” identified 485 acetylated peptides that are upregulated more than 2-fold in mouse embryo fibroblasts lacking SIRT1. 22 However, unambiguous assignment of an acetylated protein as a SIRT1 substrate is complex because SIRT1 also negatively regulates the activities of a number of protein acetyltransferases (CBP/p300, 23 Tip60, 24 Myst1, 25 and Myst2 22 ). Thus, ablating SIRT1 may increase the level of acetylation of a protein 1) because the protein is a substrate for SIRT1, 2) because the protein is acetylated by one of the protein acetyltransferases whose activity is modulated by SIRT1, or 3) for some other reason related to divergent paths by which the compared fibroblasts became immortalized in cell culture.
Biological Functions of SIRT1
In studies carried out primarily on cancer cells growing in culture, an impressive array of cellular processes has been shown to be regulated by SIRT1. These include apoptosis,26-29 autophagy, 30 cell motility, 31 drug resistance, 32 genome stability, 33 stress and drug resistance,32,34 chromatin structure and epigenetic regulation,35,36 and circadian rhythm.37,38 SIRT1 is required for the efficient differentiation of myoblasts,39,40 chondrocytes, 41 endothelial cells, 42 neural stem cells, 43 mesenchymal stem cells, 44 hematopoietic stem cells, 45 and keratinocytes. 46 In addition, whole animal studies indicate a role for SIRT1 in gluconeogenesis,47-49 metabolic syndrome,50,51 inflammation, 52 and atherosclerosis. 53
It is perhaps not surprising that so many cellular processes are dependent on SIRT1 function because the biochemical data indicate that SIRT1 has many substrates with very diverse functions. However, mice have been created with no detectable SIRT1 protein, 54 a truncated SIRT1 lacking much of the catalytic domain, 55 or a SIRT1 protein with no catalytic activity. 56 Animals homozygous for mutant Sirt1 genes are born live and can survive to old age (more than 2 years) with modest or no compromise to the plethora of cellular functions described above as dependent on SIRT1.
Mice Carrying Genetically Modified Sirt1 Genes
No SIRT1 protein is detectable in Sirt1−/− cells carrying 2 knockout alleles. 54 SIRT1-null mice develop in utero to term but have essentially 100% perinatal lethality when with an inbred genetic background. However, when carried with an outbred background, more than half of the SIRT1-null animals survive to adulthood, and some have lived for more than 2 years. One interpretation of the difference in survival with different genetic backgrounds is that the inbred backgrounds carry fixed mutations in genes that are synthetically lethal with the SIRT1-null state.
Efforts have been made to determine whether the neonatal lethality of the Sirt1−/− mice could be attributed to the failed regulation of p53, a well- established substrate of SIRT1. However, deletion of p53 in Sirt1−/− mice did not affect the Sirt1−/− phenotype,33,57 indicating that hyperactivation of p53 is not the source of the neonatal lethality of the SIRT1-null mice. Similarly, PARP has been reported to be regulated by SIRT1,58,59 but PARP deficiency does not rescue the Sirt1−/− phenotype. 60 Also, the upregulation of IGFBP1 in Sirt1−/− mice 19 appears not to be responsible for the neonatal lethal phenotype because IGFBP1 deficiency has no effect on the Sirt1−/− mice. 61 Thus, we cannot attribute the neonatal lethality of the Sirt1−/− mice to deregulation of any one of these 3 proteins.
The Sirt1−/− adult mice that survive to adulthood do suffer from a number of rather subtle phenotypes including abnormally high basal metabolic rates, 62 elevated rates of spontaneous activity, 19 decreased fertility,54,63 elevated autoimmunity,64,65 craniofacial abnormalities, 56 osteoarthritis, 66 and compromised cognition 67 and wakefulness. 68 Although diverse and large in number, these phenotypes are much less severe than might be expected if one extrapolates from the cell culture results mentioned above.
Recently, we have created a mouse line carrying a point mutation in the SIRT1 gene (Sirt1Y, formally labeled Sirt1tm2.1Mcby) that encodes a SIRT1 protein with an H355Y substitution that ablates catalytic activity. 56 Mice homozygous for this point mutation, Sirt1Y/Y, share with Sirt1−/− mice the phenotypes of small stature, elevated rates of respiration, and male infertility. However, these 2 mouse lines differ in other respects; for example, Sirt1−/− are female sterile, while Sirt1Y/Y are female fertile, and Sirt1−/− have an eyelid inflammatory phenotype, while Sirt1Y/Y mice generally do not. The difference between the phenotypes is sufficient to suggest that the SIRT1 protein may perform other functions in addition to protein deacetylation.
Confusion Surrounding SIRT1
Much of the literature involving SIRT1 is confusing and controversial. One reason is that many studies use resveratrol or SRT1720, a natural and a synthetic small molecule, respectively, which were initially reported to directly enhance the catalytic activity of SIRT1.50,69-72 The weight of evidence now indicates that neither resveratrol73,74 nor SRT172075,76 directly modulates the catalytic activity of the SIRT1 enzyme (although there may still be residual confusion regarding SRT1720 77 ). Resveratrol has been studied intensely and mimics CR in reducing the incidence and severity of a variety of experimentally induced diseases (including metabolic syndrome, cancer, and cardiovascular disease). 78 The beneficial effects of resveratrol often require SIRT1, 79 but the mechanism(s) responsible for the action of this drug remain controversial. 78
A large number of transcription factors and co-factors have been reported to be regulated by SIRT1 (Table 1), so it is surprising that microarray 80 and proteome analyses 22 of cells lacking SIRT1 showed little difference from the wild type. In fact, the literature is full of conflicting results on the effects of SIRT1 on various biological processes. For example, the tumor suppressor p53 is a well-documented SIRT1 substrate whose transcription- and apoptosis-inducing activities were first reported to be inhibited by SIRT1,26,27,81 but subsequent studies failed to find any effect of SIRT1 on p53 function.57,82 The oncogene c-myc is another SIRT1 substrate that has been variously reported to be destabilized83,84 and to be stabilized85,86 by SIRT1-dependent deacetylation.
The literature linking SIRT1 to autophagy is also illustrative of the confusion and complexity that abounds in the SIRT1 literature. In 2008, it was first reported 30 that SIRT1 activity stimulated autophagy and that autophagy was severely compromised in the absence of SIRT1. The contention that SIRT1 promotes autophagy was supported by a number of subsequent reports.87-90 Remarkably, however, several other reports have appeared showing that SIRT1 inhibits autophagy.91-93 Our own work with primary fibroblasts and neurons derived from our Sirt1−/− and Sirt1Y/Y mice showed no defect in autophagy induced by glucose deprivation (Caron et al., unpublished).
SIRT1 is a Protein Network Hub
Protein-protein interaction studies have established that SIRT1 can bind to a broad spectrum of proteins in addition to its known substrates (Table 2). In some cases, the SIRT1 protein regulates the function of the interacting partner (such as EGR1 94 and AP1 95 ), while in other cases, the interacting partner regulates SIRT1 activity (e.g., DBC196,97 and AROS 98 ). A recent meta-analysis puts SIRT1 into an exclusive class of highly networked proteins with 136 primary nodes and 4,691 second-order nodes. 99
A Partial List of Proteins That Can Bind to SIRT1 but Are Not Currently Established Substrates for SIRT1 Catalysis

Note: Those indicated in bold are cytoplasmic, while those not in bold are nuclear.
Protein association networks in eukaryotic cells are generally thought to be “scale-free” or “small-world” networks in that some highly connected nodes (hubs) can have hundreds, perhaps thousands, of interactions, while others have very few. SIRT1 is a scale-free network hub (Fig. 1), as are many of its interacting partners (p53, NF-κB, p300, c-myc, etc.). An important characteristic of a scale-free network is its robustness. 100 The random loss of a node is unlikely to have severe consequences for the network because of the low probability that any node will be a hub. Even the loss of a hub is not necessarily lethal when there are enough other highly connected nodes in the vicinity of the affected hub to carry signaling information. This kind of redistribution of information flow through the scale-free network may explain the surprising viability of animals in which the function of a hub protein has been eliminated (e.g., p53-null mice). With the loss of a hub, the functioning of a scale-free network may be compromised in cryptic and unpredictable ways. These uncertainties are relevant in established cell lines where the loss of one or more hubs may confer an advantage in vitro (e.g., resistance to apoptosis).
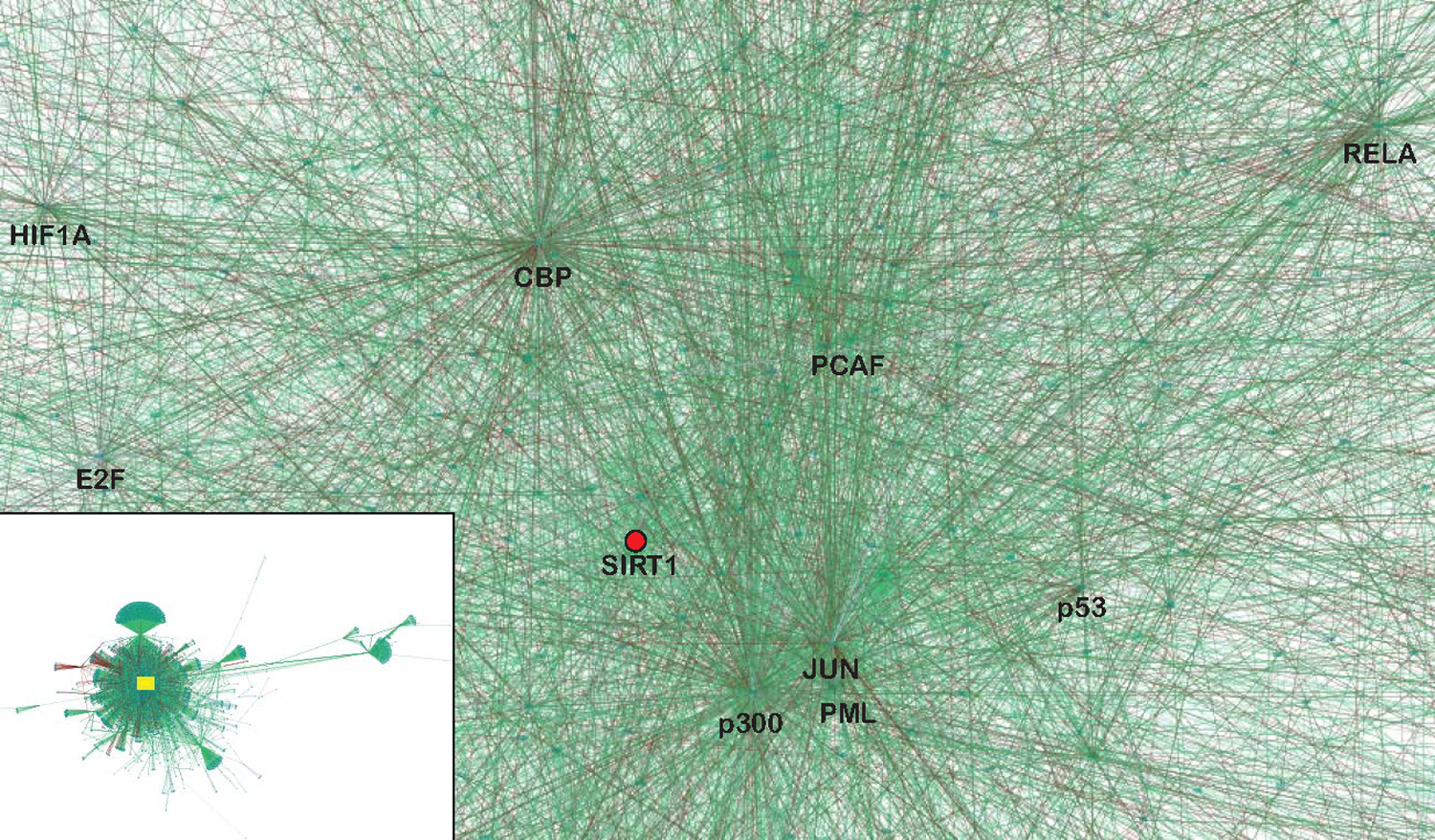
SIRT1 is a hub embedded in a network of hubs. The image depicts a portion of an edge-weighted, force-directed SIRT1 protein interaction network generated from public databases by Cytoscape. 215 Edges represented as green lines refer to interactions reported in the BioGRID database. Edges represented as red lines refer to interactions reported in the HPRD database. The SIRT1 hub is represented as a red dot. The interactions depicted are a small subset of all interactions connected to SIRT1. The entire interaction network appears in the inset, with the location of the expanded region of interest indicated by the box.
The structure of the catalytic domain of model sirtuins has been solved, 101 but computational methods applied to the full-length SIRT1 sequence indicate that the N- and C-terminal portions of the protein are largely unstructured, 102 which is an observation consistent with the idea that SIRT1 can interact with a vast array of different proteins by “coupled binding and folding.” Bioinformatic analysis of proteins found to contain large unstructured regions (designated intrinsically disordered proteins, or IDPs) has revealed that they are highly enriched in proteins that are network hubs. 103 The prevalence of IDPs as hub proteins is consistent with their property of binding with high specificity and low affinity. 104 Disordered regions of hub proteins can adopt a variety of structures depending on the identity of the interacting partner protein; one disordered peptide within the p53 protein is known to adopt a β structure when complexed with a sirtuin, a short α helix when complexed with an S100 protein, and at least 2 other stable structures when complexed with other partners. 105 The “restrained promiscuity” of hub proteins such as SIRT1 is thought to be a consequence of their disordered regions.
The SIRT1 Scale-Free Network Explains Inconsistencies
The literature is replete with studies claiming essential and critical roles for SIRT1 in various cellular processes 3 including developmental processes (see above), so it is surprising that mice lacking SIRT1 or expressing SIRT1 without catalytic activity are only modestly compromised during embryogenesis. Indeed, most of their physiological processes seem to function normally even into adulthood. There is no evidence for compensatory increases in other sirtuins that might blunt the effect of the loss of SIRT1 function. Is it possible to reconcile the observations made in cell cultures with those made in whole animals?
There are obvious potential pitfalls associated with using pharmacological and RNAi reagents with an unappreciated lack of specificity. 106 Also, ectopic expression of genes mediated by viral vectors is not without the hazard of nonspecific effects.107,108 However, we believe that the resolution to the apparently conflicting results lies with the fact that much of the published work was carried out on immortal cancer cell lines growing in culture. Each of these cell lines carries a unique collection of mutations (or epigenetic alterations) affecting such oncogenes and tumor suppressor genes such as p53, Rb, NF-κB, c-myc, and others. All of these proteins are part of the SIRT1 interaction network 99 (indeed, these proteins are both SIRT1 substrates and binding partners), and each cell line will have a different set of compro- mised proteins depending on its differentiated state and on the mutations it sustained during the progression and acclimation to cell culture. Hence, the physiological effects seen in these cells are attributed by investigators to SIRT1, but our contention is that they really derive from the combination of defects in the other proteins comprising the interacting network as well as the introduced modulation to SIRT1. Thus, the physiological effect seen following SIRT1 modulation is really a synthetic phenotype dependent on some of the other alterations carried by the cell under investigation. One can imagine that the effect of compromising SIRT1 might have no effect in a wild-type background but could either cause the activation or repression of a physiological response depending on the state of the other proteins that comprise the network. Synthetic lethality is also a likely explanation of the neonatal lethality of Sirt1−/− and Sirt1Y/Y mice when carried on inbred mouse strains.56,109
SIRT1 has already been implicated in a number of regulatory feedback loops. These include those described for regulating SIRT1 activity and c-myc,85,110 p53, 111 PPARγ, 112 and N-myc. 113 Many of the feedback loops or sources of SIRT1 regulation involve microRNAs, more than 23 of which are now reported to associate with the 1,800-nucleotide 3′ untranslated region of the SIRT1 transcript. 114
Given the large number of pathways apparently governing SIRT1 abundance and activity, it seems prudent to consider SIRT1 to be a hub in a complex scale-free network rather than a critical node in a number of overlapping, independently functioning pathways. Any change in SIRT1 or its partner proteins will have ramifications affecting all of the surrounding network components, so we can expect that the notions of “upstream” and “downstream” in a signaling pathway do not apply.
Adaptation to Stress Is Compromised in Sirt1−/− Mice
Although laboratory-reared Sirt1−/− mice do have relatively minor anatomic and physiological deficits, these animals are notably more sensitive than the wild type when confronted with various chronic perturbations. For example, mice with compromised Sirt1 genes develop emphysema at elevated rates when forced to inhale cigarette smoke,91,115 develop atherosclerosis at higher rates, 53 and are more prone to tissue destruction following exposure to ischemic insults to the heart 116 and brain. 117
Nutritional stress has been a favorite means of perturbing animals because chronic CR has well-established health benefits, and high calorie diets predispose to compromised health. When subject to CR, Sirt1+/+ mice adapt by upregulating their rates of respiration to acquire a new energy homeostatic steady state; however, the rates of respiration in CR Sirt1−/− 62 and Sirt1Y/Y 56 mice decrease in line with their reduced caloric intake, suggesting that these animals fail to adapt in the way that Sirt1+/+ mice do.
Excessive caloric intake results in obesity and predisposes to metabolic syndromes and diabetes. One report 118 suggests that liver-specific knockout of SIRT1 protects mice from the adverse effects of high calorie diets, whereas most reports indicate that elevated liver SIRT1 is beneficial51,119,120 and SIRT1 deficiency121,122 exacerbates the defective glucose homeostasis and liver steatosis induced by the high fat diet.
A parsimonious interpretation of these observations on mice carrying Sirt1 mutations is that SIRT1 accelerates the physiological adaptation to chronic stress by being a member of a network that responds to low (nonlethal) levels of chronic stress by assuming different stable states and that the efficiency with which these transitions occur is dependent on SIRT1 and, one presumes, other components of the network. The fact that SIRT1 is a key node in the complex network of interacting proteins suggests that it plays an important role in allowing cells to transition between stable states during chronic perturbation.
How might SIRT1 effect an accelerated adaptation to chronic stress? One possibility derives from the observation that many sublethal stresses result in moderately elevated levels of reactive oxygen species (ROS) derived from mitochondria. 123 One can imagine that ROS-mediated changes in cellular redox could elevate the nuclear concentration of NAD+ and hence provoke enhanced SIRT1 catalysis and/or modulate interactions with some of its network associates (indeed, H2O2 is known to modulate some of these interactions 124 ). Alternatively, ROS could activate JNK2 to phosphorylate and stabilize SIRT1, as has been described. 125
SIRT1 and Cancer
A number of original and review articles implicating SIRT1 in cancer development have appeared over the past several years. 126 There appears to be evidence supporting the idea that SIRT1 is 1) a tumor suppressor, 2) an oncogene, or 3) neither.
A role for SIRT1 in cancer is suggested by the fact that many of its substrates are established oncogenes or tumor suppressor genes, such as p53, p73, Rb, NF-κB, and c-myc. SIRT1 itself is regulated by tumor suppressor proteins HIC1, BRCA1, and p53 and the putative tumor suppressor DBC1. Many important physiological processes relevant to carcinogenesis are reported to be regulated by SIRT1. These include genome stability,33,127 immortalization and senescence,81,128-133 and apoptosis.134,135
In human malignancies, SIRT1 has been reported to be overexpressed in cancers of the colon, prostate, breast, ovary, stomach, liver, and pancreas as compared to normal tissue.136-139 In contrast, reduced expression has been reported in cancers of the colon, prostate, ovary, brain, and bladder and in BRCA1-associated breast cancers.33,136,140,141 The inconsistencies in these reports defy simple explanation.
The classic experimental test for oncogenes involves their transfection into primary or immortalized fibroblast cell lines. 142 Neither the wild-type nor mutant Sirt1 gene has any activity in these assays. 79
Genetically engineered mouse models created to overexpress or ablate SIRT1 expression have provided ambiguous evidence. On the one hand, transgenic mice that moderately overexpress SIRT1 were reported to develop slightly fewer spontaneous carcinomas and sarcomas, 143 and mice that overexpress SIRT1 in intestinal epithelium developed fewer polyps in the APCmin mouse model of colon cancer. 144 However, Sirt1−/− mice are not tumor prone, and Sirt1−/− animals do not develop more APCmin-dependent intestinal polyps or carcinogen-induced skin cancers when compared to their wild-type littermates. 79
Although it was reported87 that young Sirt1-/- animals have prostatic intraepithelial neoplasia (PIN), these mice have never been found to develop frank prostate cancer. In fact, the penetrance of PIN in these Sirt1-/- animals seems to be subject to some source of variability as yet undefined (Clark-Knowles, et al., unpublished).
Cancer is thought to proceed by the accumulation of mutations affecting oncogenes and tumor suppressor genes (many of which are members of the network immediately connected to SIRT1). The clonal cancers that develop are often characterized as having acquired an addiction to, 145 or dependence on, oncogenes. It is possible that SIRT1 might be one of the proteins to which the cancer cells become addicted since the spectrum of processes that SIRT1 influences in cancer cells in culture seems far more extensive than those that are affected by the loss of SIRT1 in whole animals. Thus, the synthetic phenotypes discussed above are simply another description of what cancer biologists call acquired dependencies.
Conclusion
The studies of SIRT1 define a field of enormous potential and of contradiction. The spectrum of diseases and physiological processes that have been reported to be modulated by SIRT1 is remarkably varied, yet the biochemical linkages between SIRT1 and the physiological readout are often obscure. Some clarity may emerge by considering SIRT1 as a member of a multicomponent protein/RNA network in which the modulation of SIRT1 has far-reaching consequences, well beyond what is normally considered in signal transduction pathways. Our interpretation of experiments carried out on whole animals is that SIRT1 functions primarily to facilitate the physiological adaptation of the organism to mild nonlethal stressors and that this function is served because SIRT1 is integral to a network of affiliate proteins and RNAs that comprise a robust network able to modulate its components to accommodate nutritional and physical perturbation. It seems likely that the modulation of SIRT1 activity by pharmacological means may achieve desirable outcomes, but the complexity of the network in which SIRT1 resides makes simple predictions imprudent.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: Work from the authors’ laboratories was funded by the Canadian Institutes of Health Research.