
Abstract
The promyelocytic leukemia (PML) tumor suppressor gene was initially identified as part of the t(15:17) chromosomal translocation associated with acute promyelocytic leukemia (APL). The PML protein is responsible for the assembly and function of characteristic nuclear domains known as PML-nuclear bodies (PML-NBs), which have been implicated in a variety of cellular functions, including growth suppression, apoptosis, and cellular senescence. PML’s many roles have been linked, at least in part, to its functional interaction with the tumor suppressor p53. It has been shown that PML favors both p53 accumulation and transcriptional activity; in turn, PML expression is directly regulated by p53, and this reciprocal regulation contributes to p53-mediated apoptosis and senescence. Nevertheless, genetic proof and in vivo assessment of the relevance of this functional crosstalk are still missing. Here we show that complete Pml inactivation, in a context of p53 heterozygosity, redistributes and expands the tumor spectrum leading to the formation of angiosarcomas and increased lymphomagenesis. Importantly, we find that Pml inactivation decreases the rate of loss of heterozygosity (LOH) in the remaining p53 allele, revealing the relevancy of p53 haploinsufficiency to tumorigenesis. Our results thus lend in vivo genetic support to the importance of the crosstalk between these two critical tumor suppressor genes.
Keywords
Introduction
PML is a pleiotropic tumor suppressor gene involved in the regulation of a variety of biological processes ranging from regulation of apoptosis, stress response, and senescence to inhibition of neoangiogenesis.1,2 The tumor suppressive function of PML was at first related to its role in the regulation of myeloid differentiation and the suppression of leukemogenesis. 2 However, subsequent studies have revealed that PML regulates multiple tumor-suppressive pathways that restrain tumorigenesis in tissues of various histological origins. Accordingly, PML expression is lost in a large set of human solid cancers. 3
It has long been known that in mice, deletion of Pml leads to impaired apoptotic response and the development of hematopoietic malignancies upon exposure to radiation or chemical carcinogens. 4
More recently, inactivation of Pml in a Pten heterozygous background has been found to promote earlier onset and accelerated progression of colon and prostate cancer, in turn unveiling a new physiological role for Pml as a negative regulator of the proto-oncogene Akt. 5 The modulatory effect of Pml on activation of the Akt signaling pathway occurs at 2 distinct levels: in addition to favoring direct Akt inhibition, Pml opposes mTOR activation in conditions of low oxygen concentrations, thus inhibiting HIF-1α synthesis and neoangiogenesis. 6 These findings have been further corroborated in studies of the compound in vivo inactivation of Pml and Tsc2, a direct negative regulator of mTOR. We have found that kidney tumorigenesis originating in Tsc2 heterozygous mice was accelerated by Pml loss through mTOR up-regulation. 7
Functionally, PML is the key component of nuclear macromolecular structures known as PML nuclear bodies (PML-NBs). In these structures, PML orchestrates a dynamic network of protein-protein interactions that define the PML-NBs as functional hubs for the regulation of diverse biochemical processes. Among the best characterized and most studied functional interactors of PML is the tumor suppressor p53.1,2
p53 is one of the most often mutated genes in human cancer, owing to its ability to regulate cell cycle checkpoints, apoptosis, and senescence. Mice suffering complete loss of p53 are viable and die mainly of thymic lymphoma by 9 months of age.8,9 By contrast, p53 heterozygous mice develop various types of sarcoma, and progression to cancer is invariably achieved through loss of function of the wild-type allele (loss of heterozygosity or selection for mutations). 8 p53 acts as a transcription factor whose activity is regulated by complex posttranslational modifications. Acetylation, phosphorylation, and ubiquitination of p53 have all been found to occur in the PML-NBs, and PML regulates these events to favor p53 stabilization and activation.1,2 For example, PML promotes p53 acetylation and favors p53-induced senescence or apoptosis upon Ras hyperactivation and DNA damage.10-12 Also, PML regulates p53 stability through regulation of p53-Mdm2 interaction. Thus, in the NBs, PML promotes p53 modifications that protect it from Mdm2-mediated ubiquitination,13-15 whereas in the nucleolus, PML sequesters Mdm2 to prevent p53 ubiquitination and degradation upon DNA damage. 16
In addition to the regulatory network orchestrated by PML to induce p53 tumor suppressive functions, PML itself is a direct transcriptional p53 target upon expression of oncogenic Ras, 11 which generates a positive feedback loop that reinforces p53 responses to promote apoptosis and senescence.
The characterization of the PML-p53 regulatory axis has been achieved through a number of elegant in vitro studies, but genetic proof of their functional interaction and its relevance in vivo are still missing. To define the relative contribution of Pml to p53 functions and their possible crosstalk, we performed an in vivo analysis of p53/Pml compound mutant mice and found that loss of Pml affects tumorigenesis induced by p53 haploinsufficiency by modifying the tumor spectrum and lowering the genetic pressure for complete p53 loss.
Results and Discussion
To gain insights into the functional interconnection between Pml and p53, we crossed p53 heterozygous mice with mice heterozygous for Pml in order to generate p53 heterozygous mutants with decreasing Pml dosage: p53+/− Pml+/+, p53+/− Pml+/−, and p53+/− Pml−/− mice. Because PML has been described as a critical regulator of p53 functions in vitro,10-12 we hypothesized that reduced Pml expression would result in decreased p53 functional output and therefore more pronounced tumorigenesis. To test our hypothesis, we performed a histopathological analysis of tumor tissues from male and female p53+/− mice with decreasing levels of Pml at time of death. As expected, all p53+/− mice of a mixed 129/Sv-C57BL/6 genetic background with wild-type expression of Pml developed sarcomas with a predominance of osteosarcomas (50% of all tumors, n = 11). Fewer mice also developed fibrosarcomas (27.3%, n = 6) and squamous cell carcinomas (22.7%, n = 5) (Fig. 1A). A 50% reduction in Pml expression levels in littermate animals of the same genetic background did not greatly modify tumor distribution driven by heterozygous loss of p53: as shown in Fig. 1, B and C, the majority of p53+/− Pml+/− mice developed aggressive sarcomas, and the most representative tumor type was again osteosarcoma (60%; n = 9), followed by fibrosarcoma (20%; n = 3). In addition, however, we found that a small percentage of p53+/− Pml+/− mice developed angiosarcoma (7%, n = 1) and lymphomas (13%, n = 2), which were not observed in the Pml wild-type context.
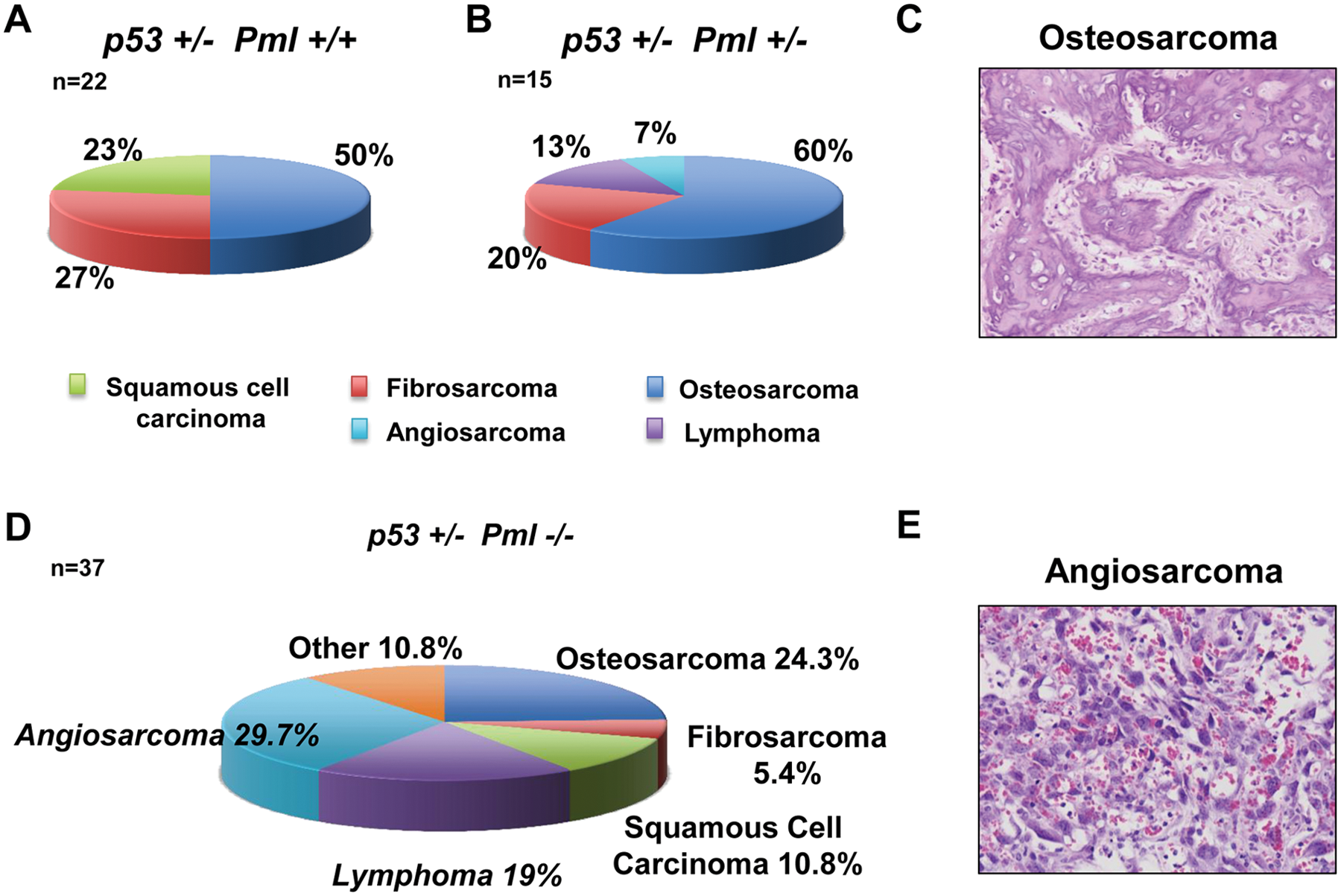
Histopathological analysis of the tumor spectrum observed in the indicated cohorts of mice.
When we analyzed the tumor burden of p53+/− mice with complete loss of Pml compared with p53+/− mice retaining Pml expression, we found a significant variation in tumor type distribution, mainly due to the high frequency of angiosarcomas (P = 0.04; Fig. 1D) and lymphomas (P = 0.03; Fig. 1E), which were never found in littermate p53+/− mice of comparable genetic background. Indeed, 11 of 37 (29.7%) p53+/− Pml−/− mice developed angiosarcomas, and 7 of 37 developed lymphomas (19%). Angiosarcomas appeared to arise at the expense of osteosarcomas and fibrosarcomas, which were reduced from about 50% in p53+/− mice to 24.3% in p53+/− Pml−/− mice (P = 0.045) and from 27% to about 5% (P = 0.017), respectively. A lower number of mice developed squamous cell carcinomas (10.8%, n = 4) and more rare types of sarcoma (10.8%, Fig. 1D). Angiosarcoma is a malignant mesenchymal tumor that arises from cells lining the walls of blood vessels: It is formed by highly proliferating endothelial cells and is rich in blood. We found mainly subcutaneous angiosarcomas developing on the head and neck area as well as the backs of the mice. In addition, 1 of 5 p53+/− Pml−/− mice developed lymphoma, a malignancy commonly found in p53 null mice, indicating that loss of Pml may modify tumorigenesis driven by p53 heterozygosity toward a phenotype normally observed upon complete p53 loss. Indeed, mice that completely lacked p53 expression died mainly of lymphoma by 9 months of age in the mixed 129/Sv-C57BL/6 genetic background in which we bred our mice, and loss of Pml did not worsen lymphomagenesis, nor did it lead to earlier sarcomagenesis or variation in the tumor-type compared with p53 null mice (data not shown). Although loss of Pml modified p53-driven tumorigenesis toward more malignant phenotypes, we found that the life span of p53+/− mice was not affected by reduced Pml levels (Fig. 2A). Also, we performed a parallel analysis of the life span of p53−/− with Pml−/− mice, and even in this setting we found no difference in overall survival (Fig. 2B).
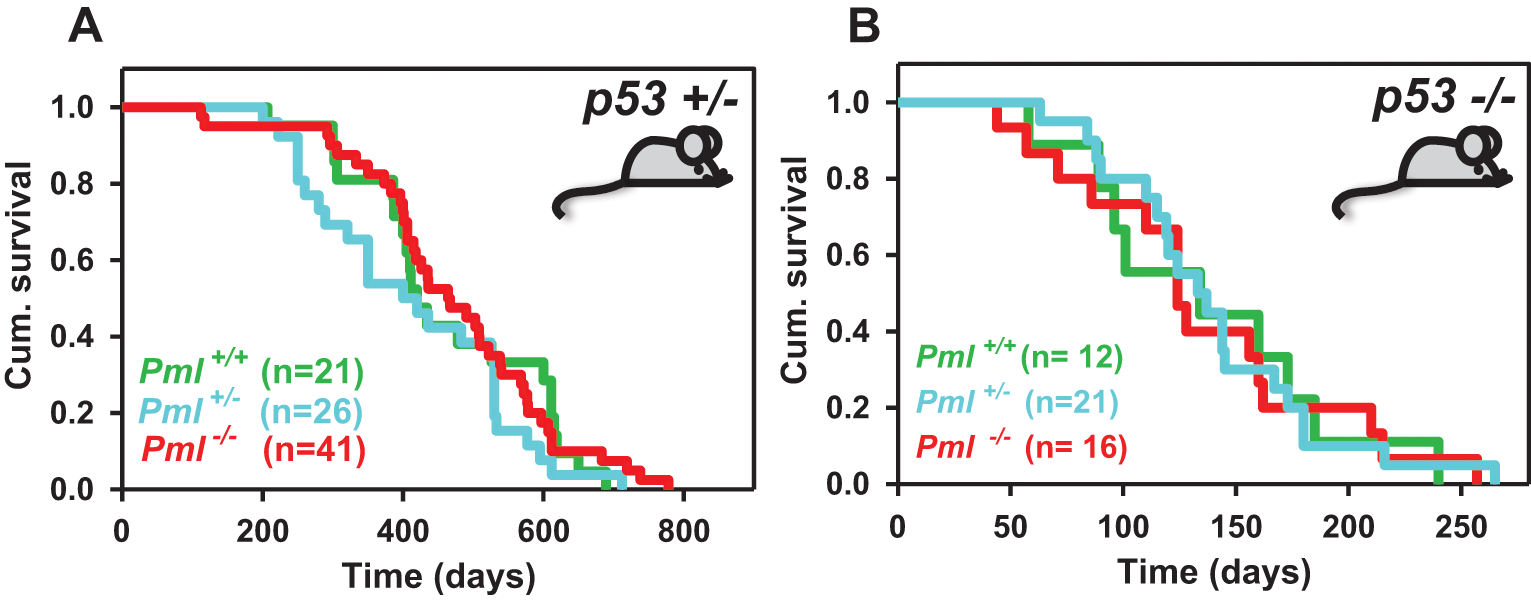
Survival analysis upon Pml and p53 inactivation. Kaplan-Meier graphs showing cumulative survival curves in p53 heterozygous mice
Critically, because tumorigenesis in p53 heterozygous mice is frequently accompanied by loss of the p53 wild-type genomic locus (loss of heterozygosity, LOH),8,17 we sought to determine whether loss of Pml expression impacted the LOH of the remaining p53 allele. To this end, we performed Southern blot analyses on DNA derived from paired tail and tumor tissues, from a number of mice with different genotypes. Our results showed that although p53+/− Pml+/+ and p53+/− Pml+/− tumors in the late stage of tumorigenesis were characterized by 80% loss of the wild-type p53 allele, in samples from p53+/− Pml−/− mice 50% of tumors retained expression of the p53 locus (Fig. 3, A and B).
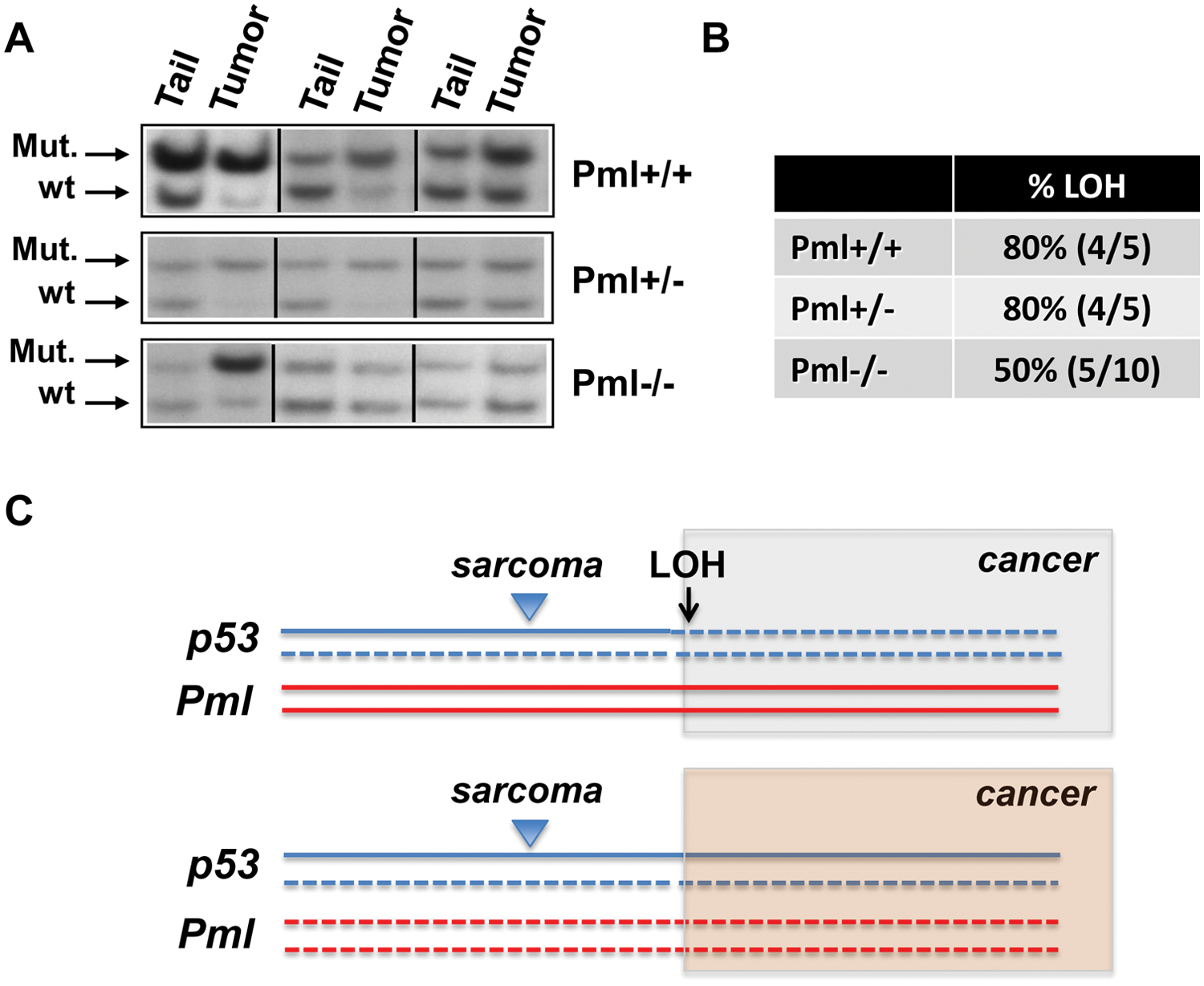
Analysis of p53 Loss of heterozygosity (LOH) on tumor tissues DNA derived from the indicated cohorts of mice.
Taken together, these results allow us to reach important conclusions on the relevance of the Pml-p53-regulated tumorigenesis in vivo.
First, we did not find significant differences in the overall survival rates of p53+/− and p53+/− Pml−/− mutant mice, but we found profound differences in tumor spectrum in the absence of Pml. These findings are in stark contrast to what we have previously observed upon crossing Pml−/− mice with Tsc2+/− mice 7 : Although in the context of Tsc2-driven tumorigenesis, loss of Pml affects tumor aggressiveness in the kidney epithelium by favoring mTOR hyperactivation and tumor acceleration, in a context of prevalent sarcomagenesis driven by p53 inactivation, loss of Pml prevalently modifies tumor identity. In this respect, we observed that loss of Pml caused 2 main phenotypes: a shift from osteo/fibrosarcomas to angiosarcomas, and the occurrence of lymphomas. As we had previously shown that PML regulates Hif1α levels and neo-angiogenesis in fibrosarcoma in hypoxic conditions,6,7 we can now speculate that loss of Pml in vivo defines a transformative step favoring the development of vascularized tumors in the context of a preexisting insult such as p53 heterozygosity (Fig. 3C). Second, we found the appearance of lymphomas in a small percentage of p53+/− Pml+/− mice, and more commonly in p53+/− Pml−/− mice. Because lymphomas are typical of p53−/− mice, we conclude that compound inactivation of p53 and Pml in the thymus may functionally mimic total p53 loss, thus promoting lymphomagenesis. Importantly, the fact that we also found lymphomas in p53+/− Pml+/− mice, although at low penetrance, suggests that heterozygous inactivation of Pml may also decrease p53 activity, thus enhancing the consequences of p53 haploinsufficiency in vivo. In turn, these findings provide further support to the importance in tumorigenesis of partial functional losses and compound haploinsufficiency of tumor suppressor genes. 18
Third, our analysis of p53 levels during late stages of tumorigenesis revealed a reduction of p53 LOH in the absence of Pml. In line with the lymphomagenesis data, the LOH analysis further suggests that the tumor suppressive role of p53 is strongly linked to Pml expression and that Pml loss causes a functional impairment to p53, thus mimicking p53 LOH (Fig. 3C). Although we have not performed a sequencing analysis to verify the occurrence of point mutations in the remaining p53 allele, we propose to exclude this possibility for 2 reasons: (1) it is widely accepted that point mutations in p53 may generate dominant negative/gain-of-function effects, thus leading to increased metastatic potential and invasiveness, 17 none of which was found in our mice; and (2) p53 mutations may lead to appearance of carcinomas rather than sarcomas 17 ; in our analysis however, we only identified sarcoma tumors.
Thus, our findings demonstrate in vivo the existence of a functional crosstalk between Pml and p53 and support a model in which their reciprocally balanced activities favor each other roles to coordinate tumor suppression.
Materials and Methods
Mice
Pml+/− and p53+/− mice used in this study were previously generated4,9. Pml+/− mice were crossed with p53+/− mice to generate all combinations of compound mutant mice. All mice were of mixed 129/Sv and C57BL/6 strains. Animals were sacrificed when moribund. All mice were cared for according to NIH-approved institutional animal care guidelines and upon approval by the Institutional Committee at the Memorial Sloan-Kettering Cancer Center, New York, NY.
Histopathology
Normal and tumor tissue samples were fixed in 4% paraformaldehyde (PFA) for 48 hours at 4°C, washed twice with PBS 1×, and transferred to 70% ethanol. Samples were embedded in paraffin, and sections 4-5 mm of thickness were stained with H&E according to standard protocols. P values were calculated by comparing the number of specific tumor types in mice p53+/− Pml−/− versus mice p53+/− Pml+/+ with Student t test.
Loss of heterozygosity (LOH) analysis
LOH of p53 allele was performed by Southern blot analysis as previously published 9 .
Footnotes
Acknowledgements
The authors would like to thank all members of the Pandolfi laboratory, in particular Andrea Lunardi and Ming Chen for insightful comments and Thomas Garvey for editing the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH/NCI grant (R01 CA-71692), awarded to P.P.P. A.P. was supported in part by the American-Italian Cancer Foundation Post-Doctoral Research Fellowship.