
Abstract
Anchorage-independent growth is the most significant hallmark of cell transformation, which has an intimate relevance to cancer. Anchorage or adhesion physically links cells to the extracellular matrix and allows the transmission of external mechanical cues to intracellular signaling machineries. Transformation involves acquiring the ability to proliferate without requiring mechanically initiated signal transduction, known as mechanotransduction. A number of signaling and cytoskeletal molecules are located at focal adhesions. Src and its related proteins, including p130Cas, localize to adhesion sites, where their functions can be mechanically regulated. In addition, the aberrant activation and expression of Src and p130Cas are linked to transformation and malignancy both in vitro and in vivo. These findings shed light on the importance of mechanotransduction in tumorigenesis and the regulation of cancer progression and also provide insights into the mechanical aspects of cancer signaling.
Introduction: Cancer from a Mechanical Perspective
Adhesion to the extracellular matrix (ECM) is critical for fibroblastic cells to execute their normal functions, including proliferation, survival, and differentiation. 1 The physiological integrity of cells in parenchymal organs is maintained by their anchorage to the ECM, and interruption of this can be directly linked to pathogenesis.2,3 For example, anchorage-independent growth is a major hallmark of cell transformation, and many cancer cells proliferate without requiring secure adhesion to the ECM. Furthermore, transformed cells typically exhibit morphological changes and abnormal migration activity. These observations suggest that adhesion-induced signal transduction is altered in malignant cells. In line with this, the regulation of proliferative signals and cell motility, whose dysregulation is implicated in cancer, 4 is closely associated with adhesion-related molecules such as integrins, focal adhesion kinase (FAK), Src family kinases (SFKs), and p130Crk-associated substrate (p130Cas).5-8
Cell-matrix adhesion is mediated by adhesion molecules that provide the basis for mechanotransduction, that is, the generation and transduction of mechanically initiated signals. 9 A variety of cell events, including growth, polarization, differentiation, and migration, involve mechanotransduction in the regulation of de novo gene expression as well as various posttranslational modifications of signaling molecules.10-14 These cellular mechanoreactions determine tissue behavior under specific mechanical environments.15-17 The “bridging function” of adhesion molecules between the ECM and cytoplasm enables cells to translate externally applied mechanical cues to intracellular signals. This likely involves cell-generated traction forces transmitted via the cytoskeleton and exerted on anchorage points to the ECM.10,15,18-20 ECM anchorage and the cytoskeletal network therefore mutually interact to maintain the physiological conditions in which cells adapt to their surrounding physical environments.
In the case of cell-cell adhesions, the homophilic binding of cadherins plays a significant role. Loss of E-cadherin expression in epithelial cells gives rise to the disruption of cell-cell adhesion and subsequently drives detachment from neighboring cells, attachment to the adjoining matrix, and motility.21,22 This acquirement of mesenchymal cell-like characteristics is referred to as epithelial-mesenchymal transition (EMT) and has been reported to promote tumor invasion and metastasis. 22 Loss of epithelial integrity, apicobasal polarity in particular, is associated with tumor progression and malignancy. 23 An appropriate balance between the physical properties of the ECM and the cytoskeleton is essential for maintaining cell morphology and polarity, which are critical elements in restraining carcinogenesis.2,11 In the complex sequence of biological events termed the “invasion-metastasis cascade,” cells in the primary tumor typically invade the surrounding ECM to reach blood vessels. They are then transported by blood flow to a foreign microenvironment, where they continue to survive. 22 Tumor cells undergo abnormal shape changes but maintain their viability and phenotypes during this process, suggesting that they are resistant to altered physical environments and that their mechanotransduction machinery is therefore modulated.
The correlation between abnormal gene expression and tumor malignancy has been reported for many adhesion and cytoskeletal proteins, including integrins, SFKs, and p130Cas,2,24 and this is in agreement with evidence implicating cell adhesion and cytoskeletal function in cancer progression. In this review, we discuss Src and its substrate p130Cas, with particular reference to their mechanical roles in cancer cells.
Src: Domain Structure and Regulation of Activity
The proto-oncogene encoding c-Src (hereafter noted as Src) is a nonreceptor tyrosine kinase and a member of the SFKs. Other SFKs include Yes, Fyn, Lyn, Blk, Fgr, Lck, Hck, and Yrk. 25 A Src molecule contains 3 conserved Src homology (SH) domains: SH1, SH2, and SH3 (Fig. 1). The N-terminal SH3 domain and the centrally located SH2 domain bind to specific proline-rich sequences and phosphorylated tyrosine (pY)–containing peptide motifs, respectively, whereas the C-terminal SH1 is a catalytic domain. Phosphorylation of the C-terminal tyrosine (noted as Y530 following the human c-Src sequence) by C-terminal Src kinase (CSK) 26 facilitates binding of the SH2 domain to the C-terminal tail, which leads to the closed molecular conformation of Src (Fig. 1) and suppression of its kinase activity.27,28 On the other hand, dephosphorylation of pY530 unlocks the intramolecular interaction between the SH2 domain and C-terminal tail (Fig. 1). This allows ATP and other adaptor proteins to bind Src and subsequently permits full activation of the protein. 29 v-Src has a genomic deletion in the C-terminal region, promoting constitutive activation and high oncogenicity. 30
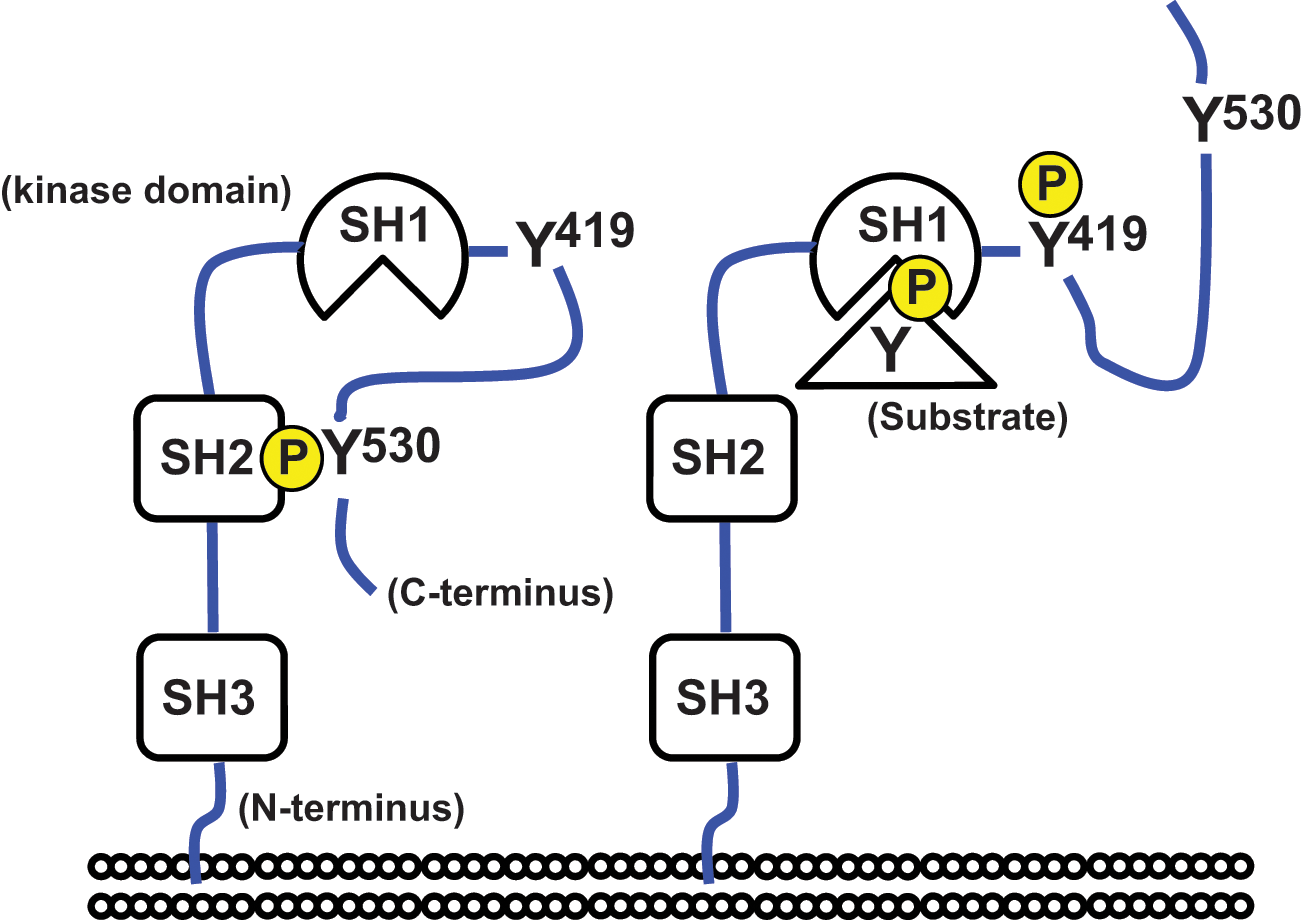
Domain structure of Src. Phosphorylation and dephosphorylation of the C-terminal tyrosine (noted as Y530) regulate the binding between the SH2 domain and the C-terminal tail, corresponding to the conversion between the closed (
Myristoylation at the N-terminus of Src tethers it to the plasma membrane. At cell-matrix contact sites, Src joins the focal adhesion complex together with integrin, actin, paxillin, FAK, and p130Cas. 31 Src phosphorylates FAK at several tyrosine residues including Y576 and Y577, 32 resulting in full activation of FAK. The Src-FAK complex regulates cell survival and proliferation via the p85/phosphatidylinositol 3-kinase (PI3K)/Akt and Grb2/Sos/Ras/Raf/MEK/ERK pathways, respectively.33,34 Furthermore, FAK functions as a scaffold for paxillin and p130Cas, which are phosphorylated by Src.35,36
Receptor tyrosine kinases (RTKs) interact with Src. Platelet-derived growth factor receptor (PVDFR) activates Src, which in turn phosphorylates PVDFR at Y934, rendering PVDF-induced mitogenesis.37-39 Src also interacts with epidermal growth factor receptor (EGFR) and phosphorylates Y845, whose phosphorylation enhances mitogenesis and transformation through ERK activation. 40
Furthermore, Src participates in signaling at cell-cell contacts. In this case, it phosphorylates p120-catenin, which interacts with and regulates the stability of E-cadherin, leading to the loss of E-cadherin function. 41
Aberrant Src Activation and Cancer Progression
The increased expression and/or activity of Src has been observed in various types of tumors.42-45 It has been reported that Src plays a central role in tumor progression by promoting cell survival, proliferation, motility, invasion, migration, and metastasis.2,46-48 Yet, it is unclear how Src is aberrantly activated in cancer. A mutation in the C-terminal tail of Src that markedly increased its kinase activity was found in late-stage colon cancer specimens, 30 suggesting the importance of the intramolecular interaction involving SH2-pY530 (Fig. 1) in cancer progression. However, mutations of Src are rarely observed in solid cancers in humans. 49 Alternatively, protein tyrosine phosphatase 1B (PTP1B), which dephosphorylates Src at Y530, 50 has been implicated in cell transformation mediated by the proto-oncogene product, ErbB2. 51 Considering that ErbB2 belongs to the EGFR family 52 and Src interacts with RTKs, cytokines, and growth factors secreted in an inflammatory microenvironment,53-55 this may contribute to Src activation during tumorigenesis.
Integrin-Mediated Src Activation and the Role of Src as a Mechanotransducer
It has been reported that upon integrin activation, FAK is recruited to the integrin complex via associations with talin and paxillin, where it is then autophosphorylated at Y397. 56 FAK Y397 phosphorylation subsequently provides a binding site for the SH2 domain of Src, which leads to its activation. 56 Integrins mechanically link the ECM to an intracellular signaling network, and therefore, Src activation via these mechanoproteins is consistent with the findings that implicate Src in cellular mechanoresponses and force transmission. 57 These studies clearly indicate that Src plays a role as a mechanotransducer. Here, we discuss the role of Src as a mechanosensor that converts physical stimuli into biochemical signals.
Using fluorescent resonance energy transfer (FRET)–based technology to monitor Src activity, Wang et al. 58 showed a rapid local activation of Src at cell-substrate contacts that were mechanically perturbed. They also demonstrated that the propagation of Src activation was in the opposite direction to the locally applied force. These observations are in agreement with Src activation by cyclic cell stretching 59 and indicate that Src is involved in mechanosensing. Moreover, we recently found that Src activity in NIH3T3 fibroblasts and mouse embryonic stem cells changes depending upon the rigidity of the substrate to which the cells adhere. 60 Considering that Src is activated upon conversion from a closed to an open conformation, which is associated with the disruption of the intramolecular interaction (Fig. 1), it appears possible that Src acts as a primary mechanosensor that is deformed by local forces, thereby facilitating downstream signaling. In agreement with this notion, it has been shown that Src can be activated rapidly (<0.3 seconds) in response to mechanical perturbation. 61 However, to mechanically dissociate the SH2 domain of Src from the sequence containing pY530 (Fig. 1), at least 2 distinct regions related to the SH2 domain and the C-terminal tail need to be anchored to the force-bearing or -transmitting units of cells, such as the actin cytoskeleton.
Although the N-terminal SH3 domain binds to cytoskeleton-associated molecules as described earlier, cytoskeletal tethering to the C-terminal region has not been demonstrated to date. In addition, it is unclear if the magnitude of mechanical force required to disrupt the SH2-pY530 interaction is within the range that can be exerted on a single Src molecule in a cell. Furthermore, we found that the increase in p130Cas phosphorylation immediately after cell stretching does not involve Src kinase activation. 62 From these perspectives, there is no solid evidence or indication that Src serves as a primary mechanosensor. By contrast, it has been reported that the Src substrate p130Cas enhances Src activity. 63 In addition, the kinase reaction of Src and p130Cas exhibits processive phosphorylation, 64 suggesting substrate-mediated regulation of Src activity. From these observations, the mechanoresponsive activity of Src may rely upon the mechanosensing role of its substrates, such as p130Cas. 62
p130Cas: Functional Subdomains
p130Cas belongs to the Cas family, which includes neural precursor cell expressed, developmentally down-regulated 9 (NEDD9); embryonal Fyn-associated substrate (EFS); and Cas scaffolding protein family member 4 (CASS4). 31 p130Cas is ubiquitously expressed, 65 and mice lacking p130Cas are embryonic lethal, showing systemic congestion with cardiac hypoplasia. 66 p130Cas comprises an SH3 domain, a proline-rich domain, a substrate domain, a serine-rich domain, and a Src binding domain, from its N- to C-terminus 31 (Fig. 2). The substrate domain (CasSD) contains 15 repeats of the YxxP motif, which provides the consensus binding sequence for SH2- and PTB-containing proteins such as Crk, Nck, and SHIP2, depending on tyrosine phosphorylation by SFKs.67-69 Among the above-mentioned functional domains in p130Cas, the N-terminal SH3 (CasSH3) and the C-terminal Src binding (CasSB) domains, but not CasSD, are reportedly required for p130Cas localization to focal adhesions. 70 As the domain name indicates, CasSB binds to SFKs, 71 whereas CasSH3 reportedly interacts with a number of proteins including FAK, Pyk2, PTP1B, C3G, and DOCK180.72-76 In contrast, only the zyxin family of proteins has been reported so far to bind to CasSD in its unphosphorylated state.77-79 With regard to the C-terminal region of p130Cas, previous studies have revealed its contribution to dimer formation and focal adhesion targeting. 70 Taken together, the functional role of CasSD appears to be specific to phosphorylation by SFKs, while the other p130Cas subdomains are responsible for protein-protein interactions that define the localization of p130Cas molecules.
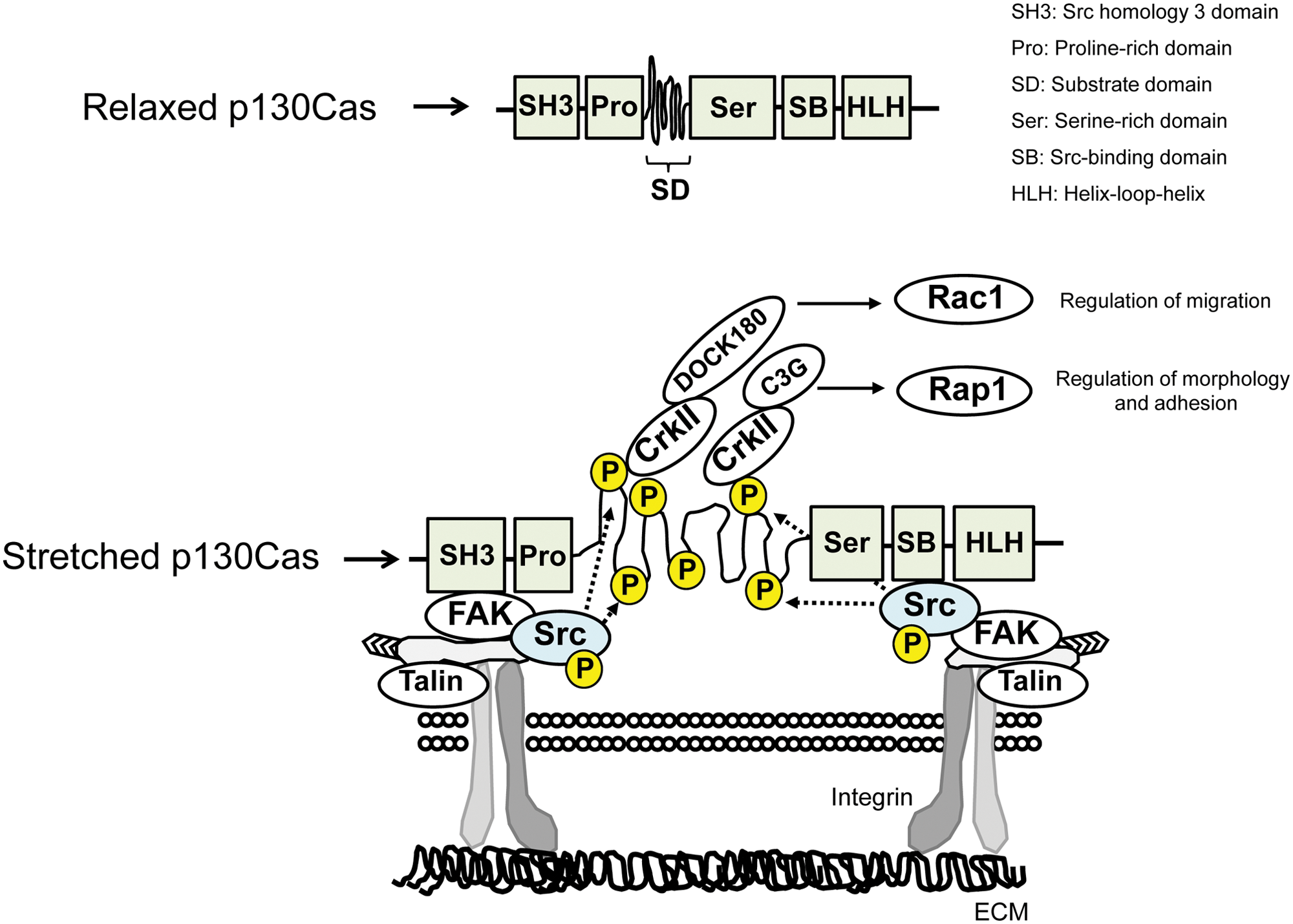
Domain structure and mechanical extension of p130Cas and its related signaling.
Mutual Regulation of the Actin Cytoskeleton and p130Cas Phosphorylation
In agreement with impaired stress fiber formation in p130Cas-deficient fibroblasts, 80 we have recently found that tyrosine phosphorylation of p130Cas mediates actin remodeling and thereby enhances megakaryocytic acute leukemia (MAL)/serum response factor (SRF) activity, promoting myogenic differentiation. 81 These findings indicate the role of p130Cas phosphorylation in regulating actin assembly/disassembly. On the other hand, we have also observed that p130Cas phosphorylation is markedly decreased when actin filaments are disrupted by treatment with cytochalasin D or latrunculin A (our unpublished observation), suggesting that the actin cytoskeleton plays a significant role in controlling p130Cas phosphorylation. Collectively, the actin cytoskeleton and p130Cas regulate each other’s function. Remodeling of the actin cytoskeleton is highly relevant to cancer, as it regulates cellular functions such as pseudopodium extension and focal adhesion turnover, which are hallmarks of cancer. 82
p130Cas as an Ion Channel–Independent Cytoskeletal Mechanosensor
Actomyosin-derived tensile force is responsible for maintaining cell shape 83 and enables cells to “probe” the mechanical properties of substrates to which they adhere. 84 Although the involvement of cell mechanosensors has been assumed, their molecular identities have remained elusive, with the exception of mechanosensitive ion channels identified in prokaryotes.85-87 By stretching detergent-insoluble cytoskeletal complexes, termed “Triton cytoskeletons,” we demonstrated that paxillin binds to the cytoskeletons in a stretch-dependent manner, suggesting the existence of an ion channel–independent cytoskeletal mechanosensor. 88 We then showed that the Triton cytoskeletons can transmit a stretch-induced signal to the small GTPase Rap1, 89 which we found acts upstream of stretch-dependent activation of p38MAP kinase. 90 Furthermore, we developed an in vitro protein extension system with which we could stretch recombinant CasSD proteins to dissect the effect of molecular stretching by eliminating the interactions of any extraneous signaling molecules. 62 Using this system, we found that p130Cas serves as a cytoskeletal mechanosensor that converts physical stimulation (stretching) into a biochemical event (phosphorylation), facilitating downstream signaling to Rap1. 62
p130Cas Expression and Cancer
It has been reported that the expression level of p130Cas correlates with the progression of certain types of cancers, such as estrogen receptor– or ErbB2-positive breast cancer, chronic myeloid leukemia, and acute lymphatic leukemia.91,92 As described earlier, p130Cas regulates signaling for survival, proliferation, invasion, and metastasis by providing a phosphorylation-dependent platform within the adhesion complex. Oncogenes such as ErbB2, Ras, B-Raf, PTEN, and PIK3CA drive cell proliferation and cancer progression via mechanisms involving p130Cas phosphorylation. 93 Mammary tumor virus (MMTV)-p130Cas mice overexpress p130Cas in their mammary glands and exhibit mammary epithelial hyperplasia. 24 The double transgenic mice generated by crossing MMTV-p130Cas and MMTV-HER2/Neu mice, whose mammary glands express exogenous p130Cas as well as the active form of ErbB2, present with an early onset of breast cancer. 24 Furthermore, silencing of p130Cas expression by RNA interference in ErbB2-transformed mammary gland cells results in impaired invasion and migration in vitro and decreased tumor growth and lung metastasis when transplanted in vivo. 3 Taken together, p130Cas is involved in the progression of malignancies, particularly breast cancer.
p130Cas in Cancer Cell Signaling and Its Mechanical Implications
p130Cas plays a significant role in Src-mediated tumorigenesis by enhancing the accumulation of the actin cytoskeleton in podosomes. 94 Src- and FAK-dependent phosphorylation of p130Cas 32 contributes to formation of the p130Cas-Crk-DOCK180 complex and consequently induces Rac1 activation at the plasma membrane, which is important for cytoskeletal remodeling and, subsequently, cell motility.35,79 Conversely, phosphorylation of CrkII, the downstream signaling molecule of p130Cas, by Abelson tyrosine-protein kinase (Abl) permits intramolecular interaction between the SH2 domain and the phosphorylated tyrosine in CrkII. This turns off p130Cas-CrkII signaling, leading to the down-regulation of cell motility.95,96 These findings indicate that the p130Cas-Crk signaling pathway is involved in the proinvasive activity of cancer cells.
It has recently been reported that matrix rigidity regulates the growth and morphogenesis of mouse epithelial cells. 97 Furthermore, lysyl oxidase (LOX)–induced collagen cross-linking and tissue stiffening have been shown to promote focal adhesions and tumor progression in vivo. 98 These observations suggest the mechanical regulation of cancer cell signaling. However, it remains unclear how tumor cells sense matrix rigidity and how cancer-related signals are subsequently induced in them. From the tissue stiffness–dependent phosphorylation of p130Cas and its co-localization with integrin β1, 98 matrix rigidity may regulate malignancy through mechanosensing by p130Cas.
Mechanical Regulation of Src and p130Cas Is Distinguished between Normal Cells and Cancer Cells
To live a normal life on earth, it is necessary to adapt to the mechanical environments, which expose us to forces derived from gravity (1 G). Furthermore, to move our body parts efficiently, our locomotive organs, that is, musculoskeletal systems, are required to respond to changes in motion and forces. Humans and animals therefore possess the molecular machinery for mechanosensing and mechanotransduction, and these include Src and p130Cas, both of which are involved in the normal differentiation of muscle81,99 as well as bone cells100,101 (Fig. 3A).
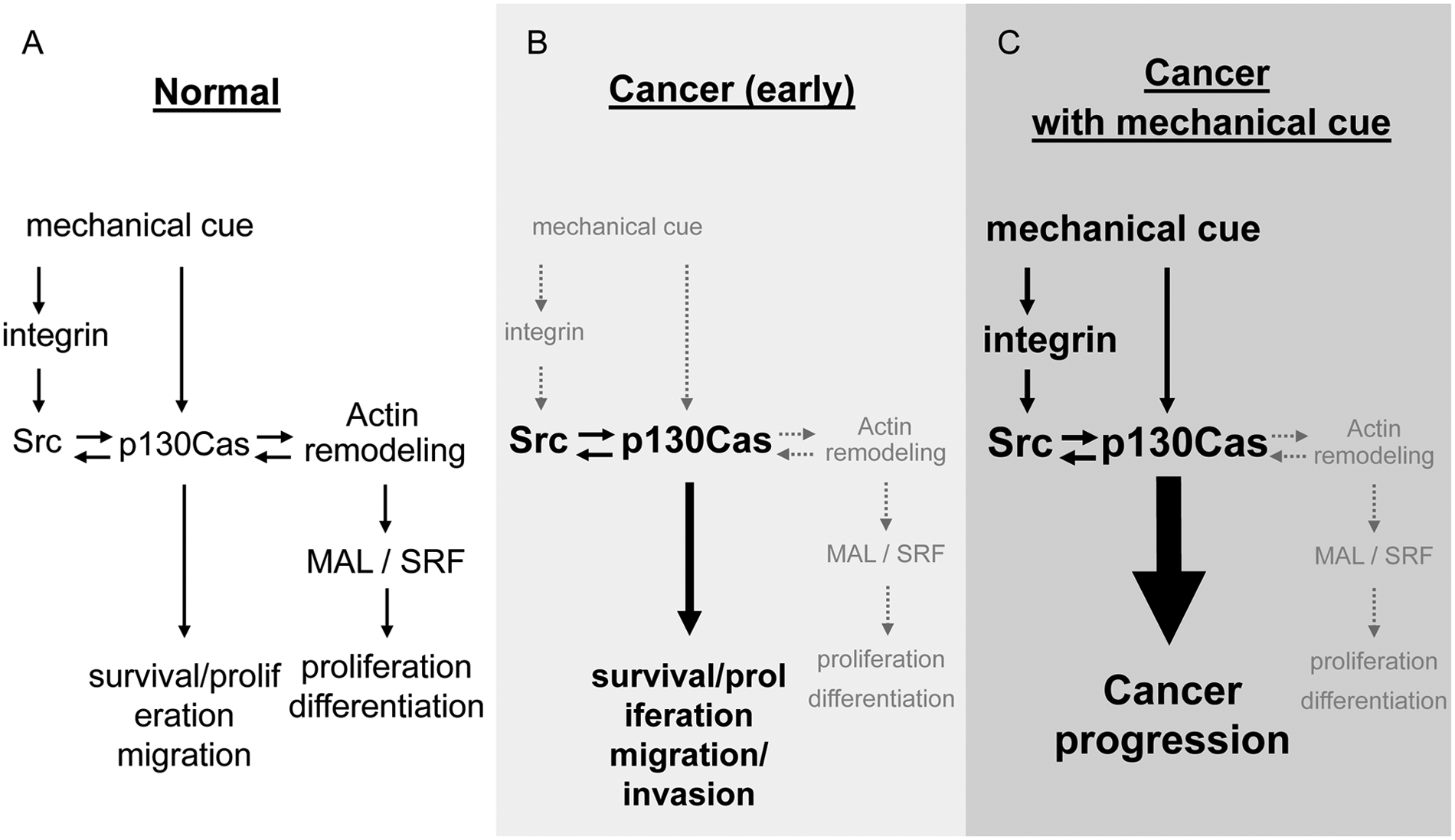
Mechanical regulation of the functions of Src and p130Cas in normal (
However, once the activity, expression, and/or function of such mechanotransducers escape mechanical regulation and get aberrant, their effects on cell survival/proliferation and migration, rather than differentiation, become overwhelming, leading to unorganized cell growth, that is, cancer (Fig. 3B). This does not appear to be limited to cell transformation by v-src. 102 For example, traction forces generated by Ras-transformed NIH3T3 cells, which carry the tumor suppressor gene p53 intact and therefore correspond to the cellular conditions of early-stage cancers, 103 are lower when compared with those generated by naive NIH3T3 cells. 104 Should the functions of Src and p130Cas in the transformed cells remain under normal mechanical regulation, their activity might be expected to decrease due to the weakened force generation and subsequent reduction in mechanotransduction. 105 However, Src activity is not decreased in Ras-transformed NIH3T3 cells (our unpublished observation), and this suggests that mechanical regulation of Src function is lost upon transformation. It follows then that cells of early-stage cancers may be less mechanosensitive in terms of Src activation (Fig. 3B). Although dysregulation of the mechanical function of p130Cas in cancer cells has yet to be established, it is possible that p130Cas is required as a co-factor for Src-induced transformation. 66 Therefore, with regard to early-stage cancers, the role of Src and p130Cas in normal and malignant cells may be distinguished by the presence and absence of mechanical regulation.
Unlike Src, whose mutation gives rise to oncogenesis, p130Cas is reportedly implicated in cancer mostly via its high expression level 31 rather than mutation. p130Cas is therefore likely to maintain its mechanosensing capability in cancer cells and tissues while remaining in the wild-type form. This notion is supported by the findings that show that traction force generation, which is the primary consequence of mechanosensing, 84 positively correlates with the cells’ metastatic potential, 106 where p130Cas also plays a role. 3 Taken together, p130Cas may facilitate downstream signaling in cancer cells when provided with enhanced mechanical cues such as matrix stiffening or increased cell-generated traction forces (Fig. 3C). It remains a mystery, however, whether mechanically regulated p130Cas function really supports cancer progression.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.