
Abstract
We have previously demonstrated proteasomal degradation of DNMT1 in mammalian cells following treatment with several DNA hypomethylating agents. Here, we demonstrate dose-dependent degradation of Dnmt1 in mouse embryonic stem (ES) cells expressing catalytic site mutant (cys-ser), confirming that the covalent bond formation between Dnmt1 and decitabine-incorporated DNA is not essential for this process. DNMT1o, the oocyte-specific isoform that lacks the N-terminal 118–amino acid domain, did not undergo decitabine-mediated degradation, which further proves the requirement of multiple domains including nuclear localization signal, KEN box, and BAH domains for this process. Analysis of glycerol density gradient fractions of micrococcal nuclease–digested nuclei showed that both nucleosomal and nucleoplasmic DNMT1 are degraded upon decitabine treatment. Among different inhibitors tested, the inhibitors of the proteasomal pathway and several protein kinases impeded decitabine-induced DNMT1 degradation. The maximal effect caused by inhibiting protein kinase C (PKC) persuaded us to investigate further its role in decitabine-mediated DNMT1 degradation. Blockage of the degradation process after treatment with rottlerin, an inhibitor of PKCδ, or after siRNA-mediated depletion of PKCδ, indicated that this protein kinase is involved in decitabine-mediated depletion of DNMT1. PKCδ interacted with and phosphorylated DNMT1 in vitro. Moreover, rottlerin inhibited both basal and decitabine-induced phosphorylation of DNMT1. These studies provide substantial evidence that decitabine-induced degradation of the maintenance methyltransferase DNMT1 does not require covalent bond formation with the substrate and also elucidate its underlying molecular mechanism.
Introduction
Methylation of cytosine in DNA at C-5 of CpG base pairs catalyzed by 3 major DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) is the most abundant epigenetic modification that usually leads to altered gene expression.1,2 The majority of cytosines in CpG base pairs are methylated on both strands in repetitive elements and in coding regions but rarely in CpG-dense regions, coined CpG islands, spanning the promoters of housekeeping genes. 2 DNA methylation is essential for mammalian development, genomic imprinting, X-chromosome inactivation, and silencing of proviral elements and retrotransposons in the genome. 3 DNA hypermethylation specific to the promoter regions is usually accompanied by recruitment of methyl-C binding proteins (MBDs), co-repressors such as histone deacetylases (HDACs), and histone methyltransferases (HMTs), resulting in silencing of the methylated genes.4,5
A hallmark of almost all types of cancer is global DNA hypomethylation as well as hypermethylation of tumor suppressor genes. 6 Unlike specific mutations, the reversible nature of DNA methylation provides an opportunity to demethylate the tumor-causing genes by DNA hypomethylating agents that can reactivate the tumor suppressor genes. Many DNA hypomethylating agents have been used to re-express the genes silenced by promoter methylation and have become a promising therapeutic strategy for certain types of cancers.7,8 5-azacytidine (5-AzaC or Vidaza) and 5-aza-2′-deoxycytidine (5-AzadC or Dacogen) are the 2 commonly used DNA hypomethylating agents. These drugs, approved by the Food and Drug Administration (FDA), have been shown to be effective in the treatment of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). 9 In addition to these drugs, several new drugs have been developed to accomplish hypomethylation of DNA and subsequent reactivation of the tumor suppressor genes. These include 5-fluoro-2′-deoxycytidine, zebularine, antisense oligodeoxynucleotides, mitoxantrone, psammaplin A, procaine, N-acetylprocainamide, procainamide, hydralazine, and (−) epigallocatechin-3-gallate. 10
We have previously demonstrated that the maintenance methyltransferase, DNMT1, is rapidly degraded by the proteasomal pathway following treatment with 5-Aza nucleotides. 11 Subsequent studies showed that proteasomal degradation of DNMT1 occurs under normal physiological conditions 12 and in response to a variety of drugs.13-16 Recently, our laboratory has shown that a quinoline-based drug, designated SGI-1027, impedes the activity of all 3 DNA methyltransferases by competing with S-adenosyl methionine and induces selective proteasomal degradation of DNMT1, causing DNA hypomethylation and re-expression of the silenced tumor suppressor genes. 17 Degradation of DNMT1 is dependent on the conserved KEN box, BAH (bromo-adjacent homology) domains, and its nuclear localization. 11 Here, we provide genetic evidence for the lack of requirement of covalent bond formation between the drug-incorporated DNA and Dnmt1 for its degradation, demonstrate the underlying mechanism by which proteasomal degradation is triggered in response to the treatment of cancer cells with DNA hypomethylating agents, and discuss its functional significance.
Results
Decitabine-induced degradation of Dnmt1 occurs in embryonic stem (ES) cells expressing the wild-type or catalytically inactive cysteine-to-serine (CS) mutant enzyme
Cysteine at the catalytic site of Dnmt1 is involved in covalent bond formation with 5-deoxyazacytidine–incorporated DNA, resulting in inactivation of Dnmt1. 18 Replacement of cysteine with serine abolishes catalytic activity as well as covalent bond formation. 3 Mouse ES cells expressing only the catalytically inactive CS mutant of Dnmt1 exhibit a defective DNA methylation pattern and are unable to differentiate. 19 To determine whether covalent bond formation between Dnmt1 and the drug-incorporated DNA is dispensable for DNMT1 degradation, we measured the Dnmt1 level in ES cells expressing the wild-type or mutant Dnmt1 following treatment with 10 µM decitabine. The results revealed a pronounced decrease in Dnmt1 levels in both wild-type and mutant cells (Fig. 1A). The basal level of Dnmt1 in the CS mutant cell line was approximately 50% of that in the wild-type cells (lanes 1 and 3) due to replacement of one allele of Dnmt1 with the CS mutant allele and deletion of the other allele. 19 The CS mutant also exhibited dose-dependent degradation of Dnmt1 with increasing concentrations of 5-AzaC (Fig. 1B). These results confirm that the covalent bond formation between catalytic-site cysteine and decitabine-incorporated DNA is not essential for degradation of Dnmt1 upon treatment with 5-azanucleosides.
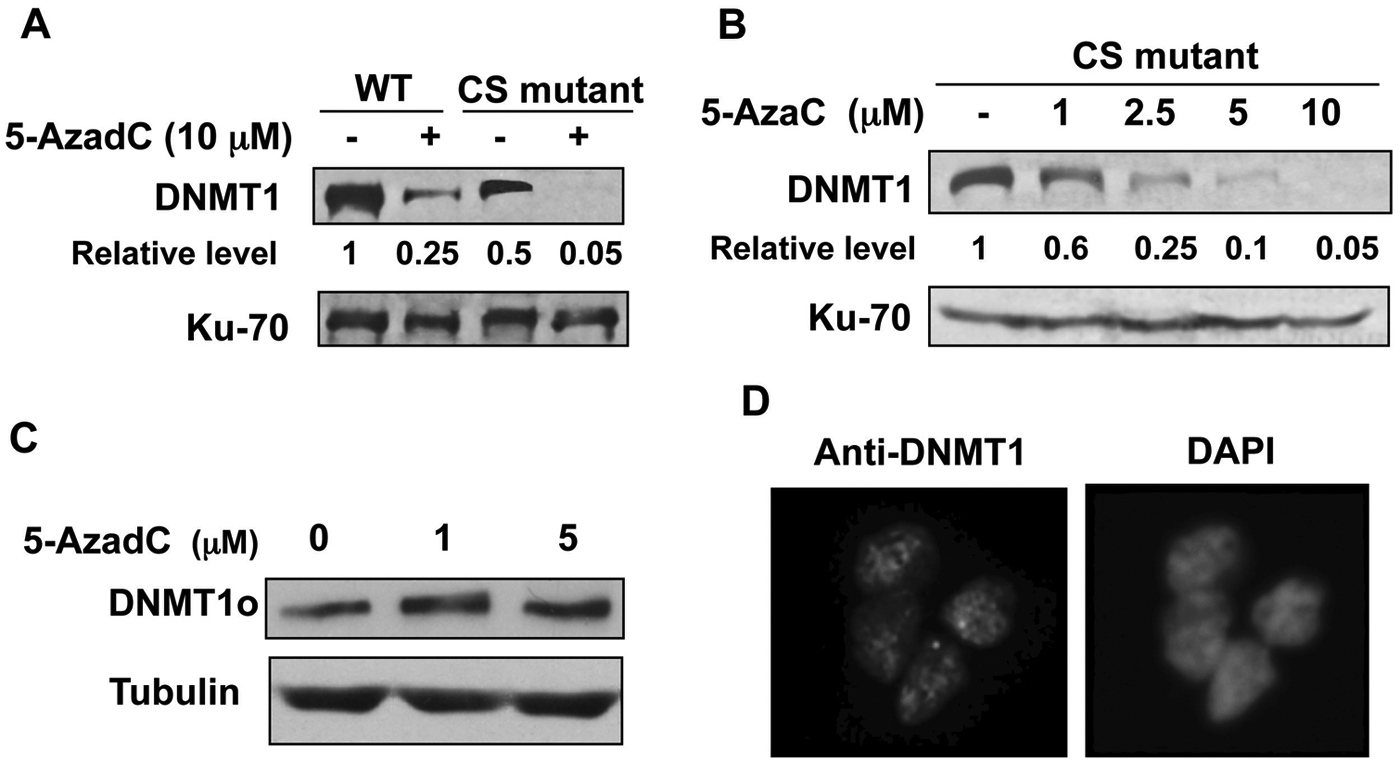
(
N-terminal domain of DNMT1 is necessary but not sufficient for decitabine-induced degradation of DNMT1
Previously, we have demonstrated that decitabine-induced degradation of the DNMT1 protein requires multiple domains of DNMT1 including nuclear localization signal, KEN box, and BAH domains. 11 It has been shown that the signal for normal turnover of DNMT1 in some cancer cells resides in its N-terminal domain harboring 118 amino acids (N118aa). 20 To determine if this holds true for drug-induced degradation of DNMT1, we used human fibroblast cells (HMT19) that stably express catalytically active DNMT1o, the oocyte-specific DNMT1 lacking N-terminal 118 amino acids. 21 Treatment of these cells with decitabine caused resistance of DNMT1o to degradation (Fig. 1C), suggesting that the N118aa peptide is required for this process. Nuclear localization of DNMT1o (Fig. 1D) indicates that the lack of decitabine-induced degradation of DNMT1o is not due to its inability to translocate to the nucleus. Next, we investigated whether the N-terminal domain harboring 118 amino acids (N118aa) can undergo drug-induced degradation. For this purpose, cell lines (H293T and HCT) expressing Flag-tagged N118aa were treated with different doses of decitabine. As observed with DNMT1o, the level of N118aa was not affected by the inhibitor, whereas the endogenous full-length DNMT1 (DNMT1-WT) level was dramatically reduced in both cell lines (Fig. 2A and 2B). These results demonstrated that neither N118aa nor DNMT1o could undergo drug-induced degradation. Because proteasomal degradation of DNMT1 occurs in the nucleus, 11 we next determined the subcellular localization of N118aa-Flag by fluorescence microscopy. Co-localization of N118aa (red) with DAPI (blue) showed that this polypeptide is predominantly localized in the nucleus (Fig. 2C). Hence, the inability of N118aa to undergo degradation is not due to its retention in the cytoplasm. Taken together, these results prove that decitabine induces degradation of only full-length DNMT1.
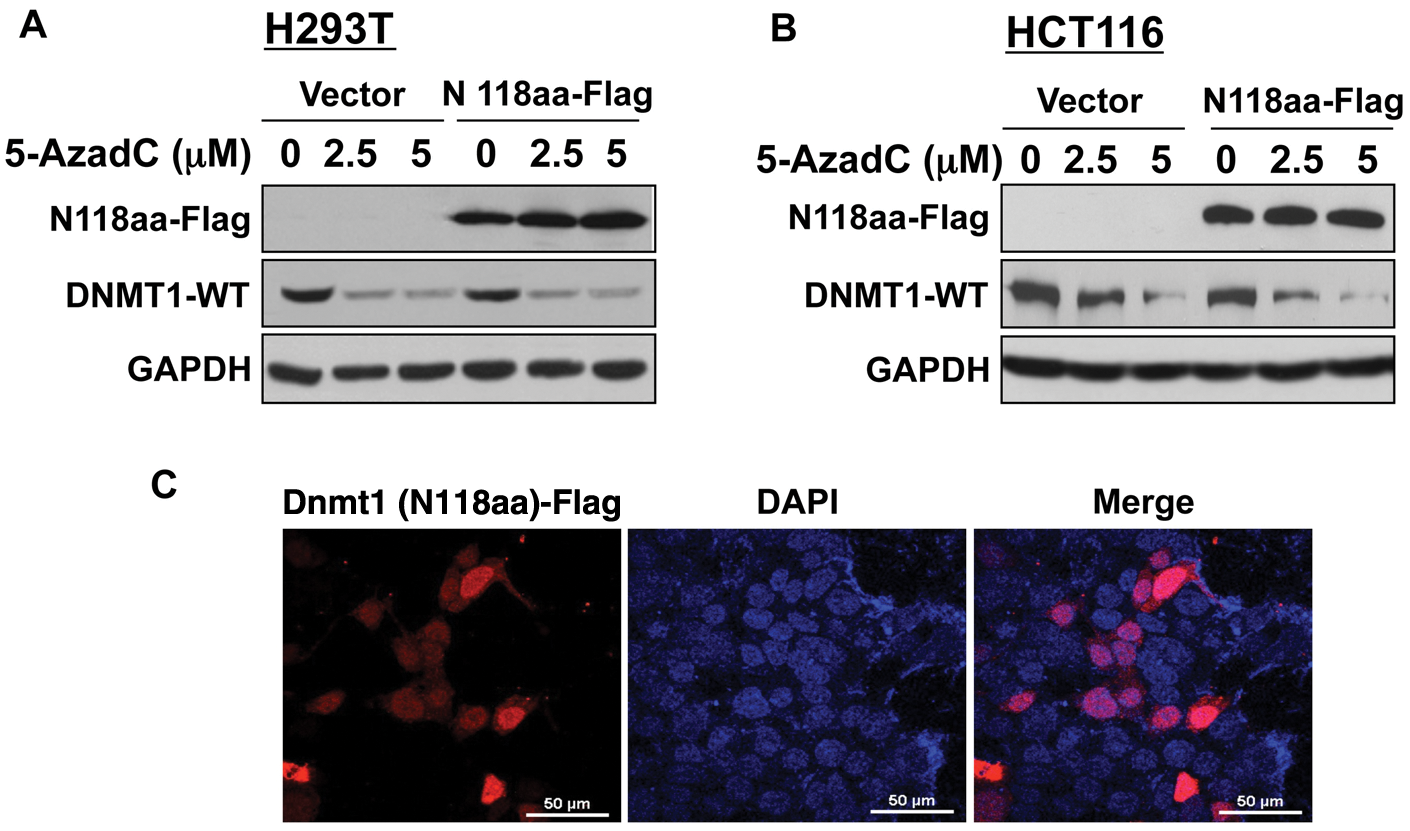
N-terminal 118 amino acids is necessary but not sufficient for decitabine-induced degradation of Dnmt1. H293T and HCT116 cells were transiently transfected with a Flag-tagged N-terminal 118–amino acid sequence of the Dnmt1 protein. After 24 hours, cells were split and treated with decitabine or left untreated for 12 hours. (
Decitabine treatment does not alter nucleosomal distribution of DNMT1
It has been hypothesized that the covalent bond formation of DNMT1 with decitabine-incorporated DNA results in its association with polynucleosomes as opposed to its association with the nucleoplasm and oligonucleosomes in untreated cells.13,15 To determine if this indeed occurs after treatment with the inhibitor, we fractionated nucleosomes obtained after partial MNase digestion of the nuclei isolated from control and decitabine-treated HCT116 cells by glycerol density gradient centrifugation and subjected the collected fractions to Western blot analysis (Fig. 3A and 3B, top panel). Nucleosome profile in each fraction was determined by fractionating purified DNA in agarose gel (Fig. 3A and 3B, bottom panel). The results showed that DNMT1 was predominantly associated with oligonucleosomes (fraction #12-14) in control cells, although a minor population was detected in the nucleoplasm (#18). Notably, the DNMT1 fractionation profile of decitabine-treated cells was very similar to that of the untreated cells (Fig. 3B, top panel), but its level was markedly reduced in all fractions including the peak at #13. These results clearly demonstrated that treatment of HCT116 cells with the DNA hypomethylating agent did not cause a shift in the association of DNMT1 from oligonucleosome to polynucleosome fractions. Like DNMT1, DNMT3B was also predominantly associated with oligonucleosomes. In contrast, DNMT3A was detectable in the nucleoplasm and in almost all nucleosomal fractions, although it was enriched in the oligonucleosomes. DNMT3A level was also reduced upon decitabine exposure. These results established that decitabine treatment does not cause a shift in the association of DNMT1 from oligonucleosomes to polynucleosomes.
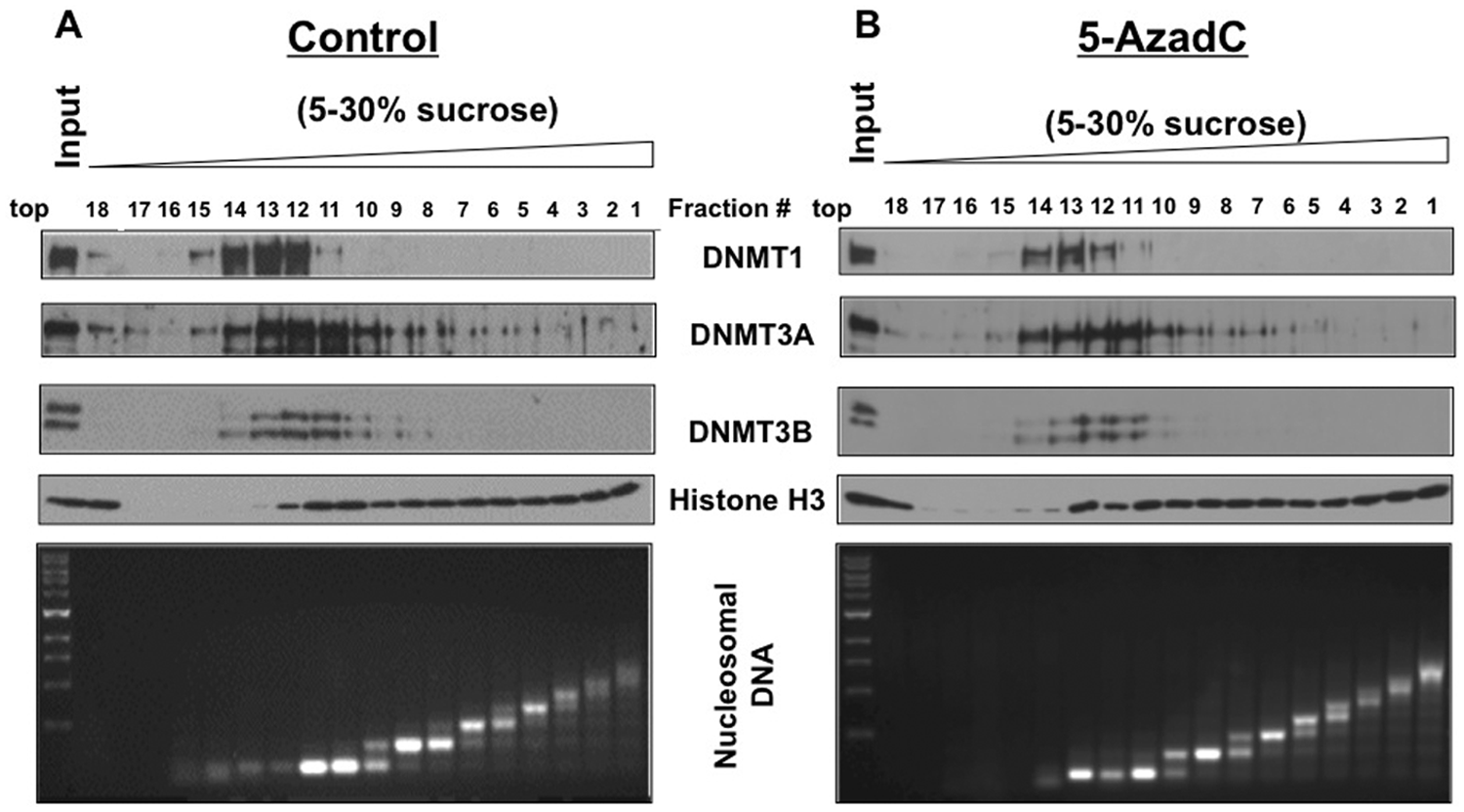
DNMT1 is predominantly associated with oligonucleosomes in HCT116 cells, and decitabine treatment causes depletion but does not shift it to polynucleosomes. (
Inhibitors of proteasome, PI3K, and PKC can impede decitabine-induced degradation of the DNMT1 protein
Based on the present results and past data, we hypothesized that proteasomal degradation of DNMT1 is induced by activation of one or more signaling pathway(s) following treatment of cells with 5-AzaC or 5-AzadC. To identify such pathways, HCT116 cells were treated with inhibitors of different signaling pathways before decitabine exposure followed by Western blot analysis. As demonstrated previously,11,17 treatment of HCT116 cells with decitabine markedly reduced the DNMT1 level, which could be blocked by MG-132, a proteasomal inhibitor (Fig. 4A). Among different inhibitors tested, only staurosporine (a broad-spectrum PKC inhibitor), LY294002 (a PI3K inhibitor), and genistein (a tyrosine kinase inhibitor) 22 could prevent DNMT1 depletion by 90%, 80%, and 70%, respectively, whereas inhibitors of MAPK signaling (PD98059, U0126, SPG00125, SB203580) and phosphatases (okadaic acid) could not restore the DNMT1 level. Ro-318220, another broad-spectrum inhibitor of PKC, could also block both 5-AzadC– and 5-AzaC–induced DNMT1 degradation in HCT116 cells (Fig. 4B) and in other cell lines like RKO (~40%-60%) (Fig. 4C) and Hep3B (100%) (Fig. 4D). The inhibitor by itself did not significantly affect the DNMT1 level. These results suggest that at least one of the isoforms of PKC is involved in triggering proteasomal degradation of DNMT1 by nucleoside analogs.
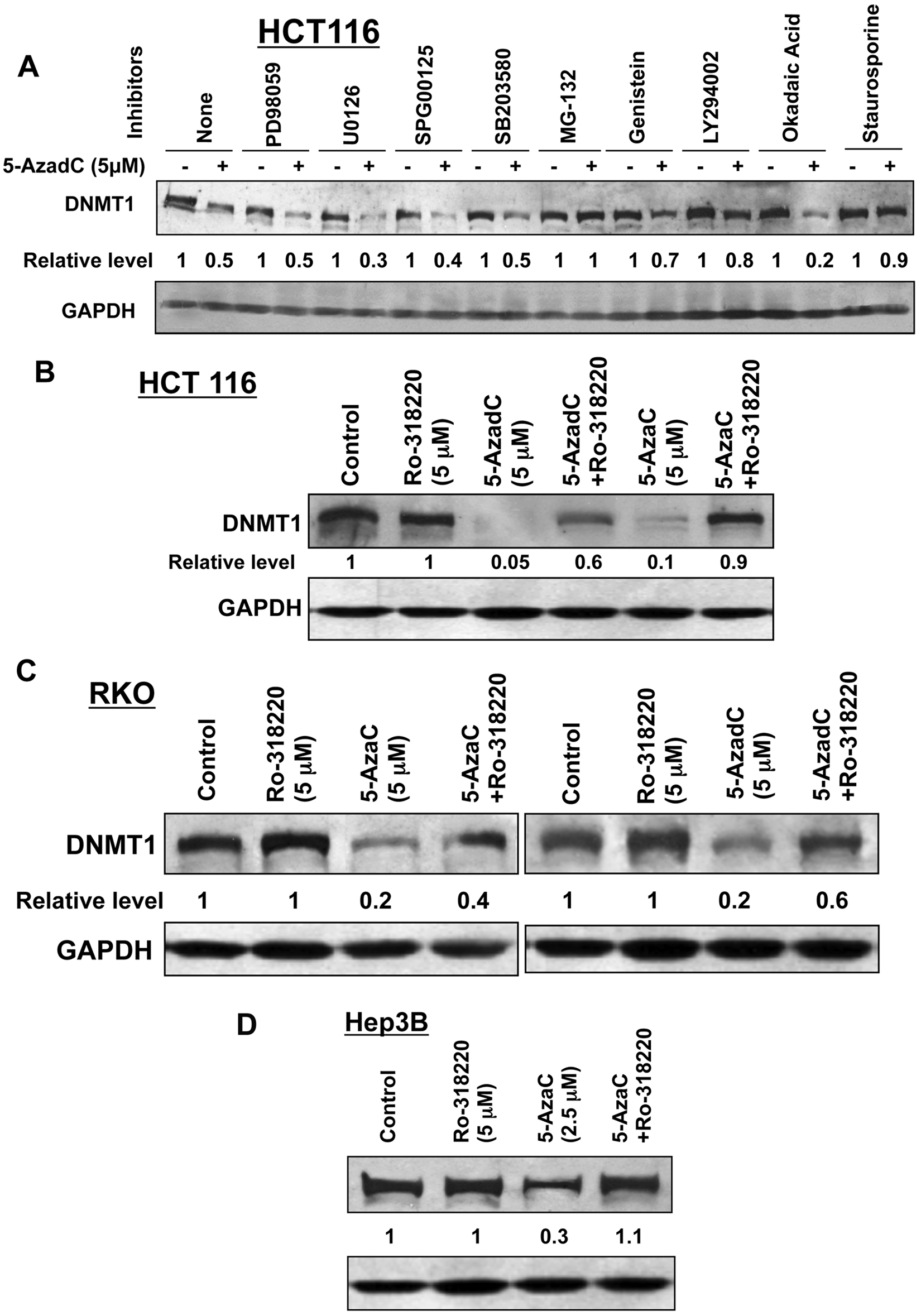
Decitabine-induced degradation of DNMT1 is blocked by protein kinase C inhibitors. (
PKCδ is involved in decitabine-induced degradation of DNMT1
Next, we sought to identify the specific isoform of PKC involved in DNMT1 degradation by pretreating cells with isozyme-specific inhibitors. Rottlerin, an inhibitor of PKCδ,23-26 was as effective as MG-132, staurosporine, and Ro-318220 in blocking decitabine-induced degradation of DNMT1 in HCT116 (Fig. 5A) cells. In contrast, G06976, a selective inhibitor of Ca2+-dependent PKCs (PKCα), was not effective in DNMT1 degradation. This observation suggests the probable involvement of PKCδ in drug-induced depletion of DNMT1 in human cancer cell lines.
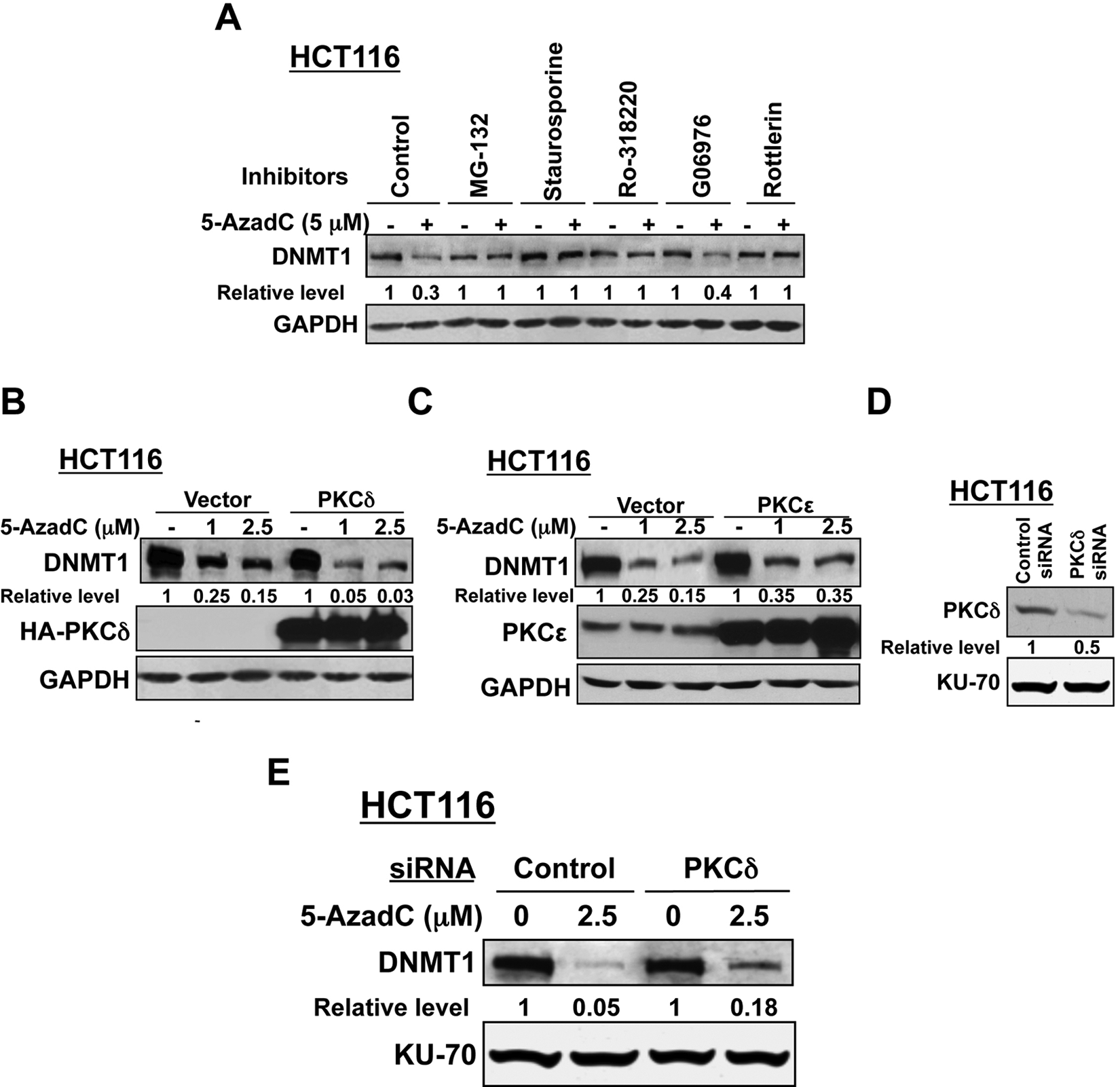
(
Because rottlerin is not a highly specific inhibitor of PKCδ, we used an alternate approach to confirm the role of PKCδ in maintaining the DNMT1 levels after decitabine exposure. For this purpose, we used cells that overexpress and underexpress the protein kinase. In vector-transfected HCT116 cells, DNMT1 level was reduced by 75% and 85% following treatment with 1 and 2.5 µM decitabine, respectively, for 12 hours, whereas DNMT1 level was almost completely depleted in PKCδ-expressing cells under this condition (Fig. 5B). In contrast, PKCϵ overexpression did not facilitate inhibitor-induced DNMT1 degradation compared to vector-transfected cells (Fig. 5C). Ectopic expressions of PKCδ and PKCϵ were confirmed by Western blotting. The role of PKCδ was further substantiated by knocking down its expression by transient transfection of HCT116 cells with siRNA that reduced its expression by approximately 50% (Fig. 5D). As expected, DNMT1 level in PKCδ-depleted cells was significantly higher than that in cells transfected with control siRNA followed by decitabine exposure (Fig. 5E).
The observation that PKCδ, a serine-threonine kinase, facilitates degradation of DNMT1 prompted us to investigate whether DNMT1 is phosphorylated at its serine moieties. To test this hypothesis, the DNMT1 protein was immunoprecipitated from HCT116 cells treated with MG-132 alone or in combination with 5-AzaC and subjected to Western blotting with anti-phosphoserine and anti-DNMT1 antibodies, respectively. The results showed that the DNMT1 protein was indeed phosphorylated at its serine residues (Fig. 6A), with approximately a 2-fold increase in phosphorylation in response to decitabine treatment. Further, phosphorylation of recombinant DNMT1 in vitro by recombinant PKCδ substantiated the notion that the maintenance DNA methyltransferase is a direct substrate of this kinase (Fig. 6B).
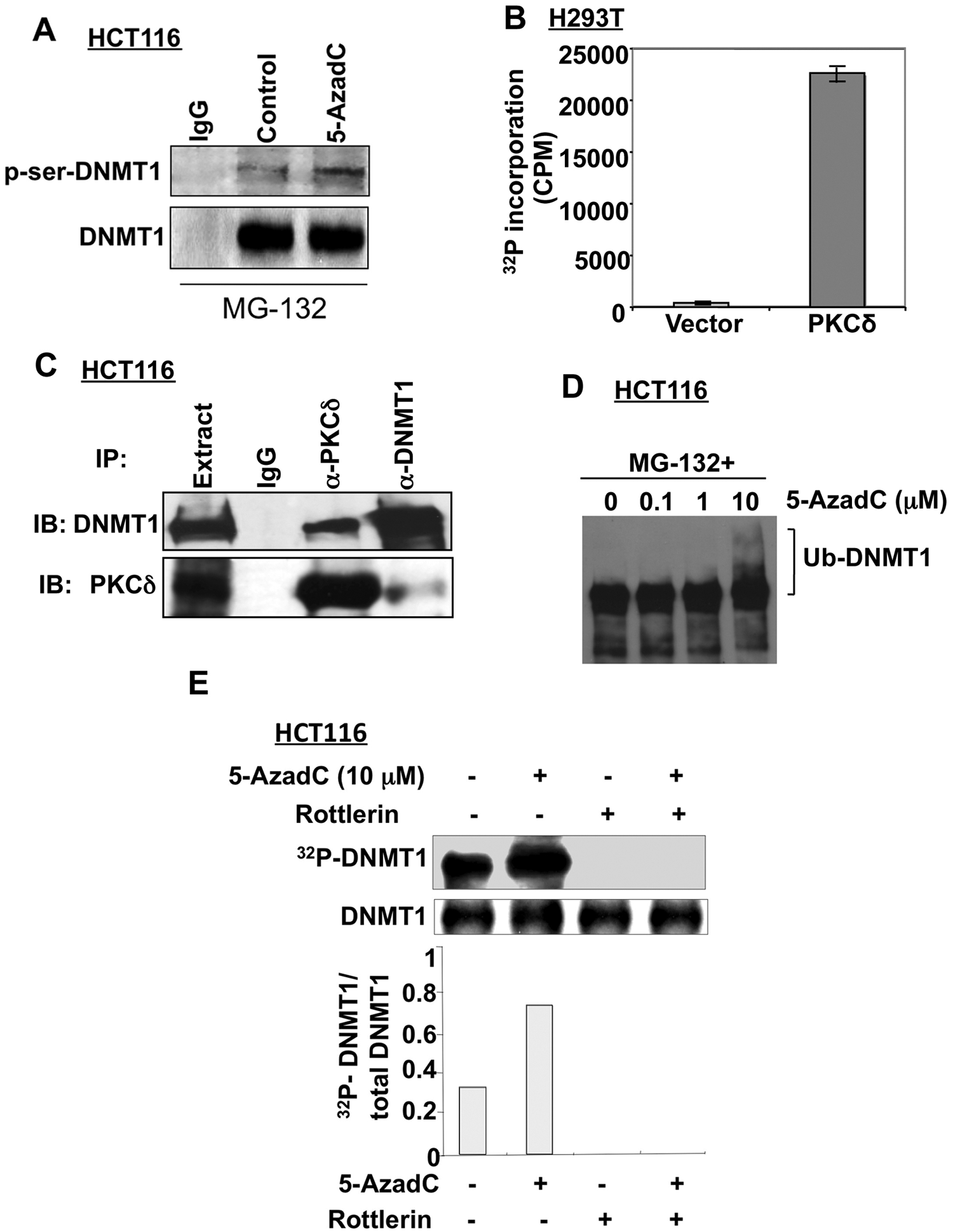
Decitabine treatment facilitates DNMT1 phosphorylation that can be blocked by PKCδ inhibition. (
We then determined if these 2 proteins interact in vivo. Co-immunoprecipitation assay showed that the DNMT1 antibody pulled down both DNMT1 and PKCδ, albeit at different levels (Fig. 6C). Conversely, anti-HA antibody specific for ectopic PKCδ precipitated both proteins, reinforcing the inference that these 2 proteins associate in vivo. The absence of detectable DNMT1 and PKCδ in the proteins immunoprecipitated by control antibody further proved the specific interaction of these 2 proteins.
Next, we investigated whether DNMT1 was phosphorylated by PKCδ and whether the phosphorylation level was altered upon decitabine treatment. To address this possibility, we first optimized the conditions under which DNMT1 did not undergo degradation after decitabine treatment. Indeed, pretreatment of HCT116 cells with MG-132, a specific proteasomal inhibitor, prevented DNMT1 degradation in the presence of 10 µM decitabine and resulted in the formation of high molecular weight DNMT1, which is probably due to accumulation of the ubiquitinated protein 11 (Fig. 6D). The phosphorylation status of DNMT1 was analyzed by pretreating the cells with MG-132 and rottlerin for 30 minutes followed by incubation with decitabine and 32P-labeled orthophosphate for 1 hour to label the phosphoproteins. DNMT1 was then immunoprecipitated, separated by SDS-PAGE, transferred onto the nitrocellulose membrane, and subjected to autoradiography followed by Western blot analysis. The results showed that phosphorylation of DNMT1 increased approximately 3-fold after decitabine treatment without significant change in its protein level (Fig. 6E). Inhibition of both basal and drug-induced phosphorylation of DNMT1 by rottlerin suggested the involvement of PKCδ in DNMT1 phosphorylation. Taken together, these results indicate that the hyperphosphorylation of DNMT1 in response to decitabine treatment probably induces its proteasomal degradation.
Discussion
The FDA-approved nucleoside analogs, 5-AzaC and 5-AzadC (decitabine), have been used for epigenetic therapy of MDS, AML, and lately in various solid tumors.27-33 Despite their wide use as DNA hypomethylating agents, their exact mode of action has not been completely elucidated. At low concentrations, these DNA hypomethylating agents cause DNA demethylation, thereby reactivating silenced tumor suppressor genes, whereas higher doses of these agents induce apoptosis through DNA double-strand breaks.34,35 It has been reported that decitabine induces cell death via P53-mediated apoptosis due to activation of ATM/ATR cell cycle checkpoint pathways in response to genomic DNA damage. 36 Although a close relationship appears to exist between the biological activity of these aza compounds and their incorporation into DNA and/or RNA, we previously demonstrated for the first time that proteasomal degradation of DNMT1 in the absence of DNA synthesis is a key mechanism by which DNA hypomethylating agents reactivate tumor suppressor genes silenced by promoter methylation. 11
Although rapid and selective proteasomal degradation of the maintenance DNA methyltransferase by the proteasomal pathway is a major contributing factor in the suppression of promoter methylation by 5-AzaC or 5-AzadC, the machinery involved in the initiation of the degradation process has not been delineated. The data presented here have shed light on this mechanistic aspect. More specifically, we have shown that 1) only the full-length DNMT1 protein (wild-type or catalytically inactive) is targeted for proteasomal degradation; 2) the N-terminal 118–amino acid domain of DNMT1 is necessary but not sufficient for the degradation process; 3) among several isozymes of PKC family members, PKCδ is specifically involved in decitabine-induced degradation of DNMT1; 4) PKCδ physically interacts with DNMT1 both in vivo and in vitro and phosphorylates DNMT1 that is facilitated in the presence of the DNA methylation inhibitors; and 5) phosphorylated DNMT1 is targeted to the ubiquitination machinery for its rapid degradation. A long-standing contention has been the need for covalent bond formation between DNMT1 and decitabine-incorporated DNA to promote inhibition of the methyltransferase activity causing DNA hypomethylation. The present study has provided the first genetic evidence to prove that covalent bond formation between Dnmt1 and DNA is not essential for decitabine-mediated degradation of Dnmt1 in ES cells.
The PKC family of serine/threonine kinases plays a central role in diverse cellular processes including proliferation, differentiation, cell cycle regulation, invasion, migration, apoptosis, tumorigenesis, and chemoresistance. 37 Among 12 PKC family members, PKCδ, a novel PKC isozyme, has been well characterized with respect to its function as a tumor suppressor.38,39 Further, its involvement in apoptosis had been well established. Previous studies have demonstrated that following exposure to the apoptotic inducer Ara-C, PKCδ was translocated to the nucleus and caused phosphorylation of lamin B, thereby predisposing it to caspase-6–mediated proteolytic degradation. 40 It is also noteworthy that PKCδ-mediated phosphorylation can inhibit nuclear DNA-PK (DNA-dependent protein kinase), an essential enzyme for the DNA double-strand break repair. 41 Furthermore, several other proteins such as hRad9, c-abl, Lyn, p73β, and histone H2B are also known to be phosphorylated by PKCδ in response to DNA damage and concomitant apoptosis.42,43 The PKCδ-mediated phosphorylation of DNMT1 both in vitro and in vivo is consistent with these observations. DNA damage occurring in cancer cells and the known translocation of PKCδ to the nucleus under this condition 42 further support the notion that this protein kinase plays an important role in the degradation of DNMT1 in response to treatment with DNA hypomethylating agents. Although rottlerin may not be a specific inhibitor of PKCδ, extensive transfection studies involving overexpression and underexpression of this protein kinase and other compelling data have demonstrated that it plays a key role in the decitabine-induced degradation of DNMT1.
Because we have previously shown that the DNA hypomethylating agents promote DNMT1 degradation in cancer cells of different origins,11,17 we used mostly colon cancer cell lines for this study, assuming that similar mechanisms exist in other types of cancer cells. It is conceivable that other agents exhibiting more potency and less toxicity than the conventional DNA hypomethylating agents can also activate PKCδ-mediated phosphorylation and degradation of DNMT1, resulting in promoter demethylation, re-expression of silenced tumor suppressor genes, and ultimately tumor regression. We cannot, however, rule out the possibility that another protein kinase may also be involved in the phosphorylation of DNMT1, causing its degradation via a different signaling mechanism. Nonetheless, this study provides convincing evidence that phosphorylation of DNMT1 by PKCδ facilitates rapid degradation of DNMT1 upon exposure to 5-AzaC/decitabine and that DNMT1 depletion is not dependent upon covalent bond formation between DNMT1 and drug-incorporated DNA.
Materials and Methods
Antibodies
The antibodies used in this study were obtained from the following sources: anti-DNMT1 (sc-10222), anti-PKCδ (sc-213), anti-PKCϵ (sc-214), and anti-actin (sc-8432) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-DNMT1 (1037-1386) (BioAcademia, Osaka, Japan); anti-PCNA (14.6748.81) (eBioscience, San Diego, CA); anti-histone H3 (ab1719) (Abcam, Cambridge, UK); anti–Ku-70 (N3H10) (Lab Vision, Fremont, CA); anti-tubulin (2148) (Cell Signaling Technology, Danvers, MA); anti-GAPDH (MAB374) (Chemicon, Temecula, CA); anti-HA (Covance, Princeton, NJ); and anti-Flag M2 (F3165) (Sigma, St. Louis, MO). Anti-Dnmt3a and anti-Dnmt3b antibodies were raised in our laboratory, as described previously.44,45
siRNAs
The PKCδ siRNA smart pool and scrambled siRNA were obtained from Santa Cruz Biotechnology.
Inhibitors
PD98059, U0126, SP600125, SB203580, G06976, Ro-318220, MG-132, genistein, Ly294002, okadaic acid, staurosporine, and rottlerin were purchased from Calbiochem, San Diego, CA. 5-AzaC and 5-AzadC were obtained from Sigma.
Construction of plasmids
For constructing pcDnmt1–N-118aa–Flag, the N-terminal domain of the Dnmt1 protein harboring 118 amino acids (N-118aa) was PCR amplified from pcDnmt1-FL-Flag 11 using the following primers, N-118-HindIII-F: 5′-ttaaagcttATGCCAGCGCGAACA-3′, and N-118-XbaI-R: 5′-aatctagaGTCCTTGGTAGCAGCC TCC-3′, and cloned at the Hind III/Xba I sites of p3XFlag-CMV-14 (Sigma). The plasmid was sequenced to confirm its authenticity, and expression of the protein was confirmed by Western blot analysis using anti-Flag and DNMT1 antibodies. Expression vector plasmid of PKCδ (HA-tagged) was a generous gift from Dr. I. Bernard Weinstein (Columbia University, New York, NY). PKCϵ wild-type expression vector (Ex-A1280-MO2) plasmid was purchased from Genecopoeia (Rockville, MD).
Cell culture
Human colon carcinoma cell lines (HCT116 and RKO), hepatocellular carcinoma cell lines (Hep3B), and embryonic kidney cell lines (H293T) were obtained from ATCC (Manassas, VA) and cultured according to the supplier’s protocol. Mouse ES cells (WT and CS mutant of Dnmt1) and human fibroblast cells (HMT19 that overexpress DNMT1o) were generously provided by Drs. Masaki Okano (RIKEN, Wako, Japan) and Paula Vertino (Emory University, Atlanta, GA), respectively.
Treatment of cells with DNA hypomethylating agents and inhibitors of different enzymes
Exponentially growing cells were pretreated with the inhibitors for 30 minutes followed by exposure to 5-AzaC and 5-AzadC at indicated concentrations for different time periods. Control cells received vehicle (DMSO) only.
Western blot analysis
Whole cell extracts were immunoblotted with different antibodies, as indicated in the text and/or figures. The signal was developed with ECL 9 after incubation with appropriate secondary antibodies.
Transient transfection and RNA interference assay
H293T and HCT116 cells were seeded on the day before transfection and were transfected with pcDnmt1–N118aa–Flag, HA-tagged PKCδ, PKCϵ expression vectors, or respective empty vectors using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Cells were equally distributed into 3 plates 24 hours after transfection and 12 hours later were treated with decitabine (1 and 2.5 µM) for an additional 12 hours. Whole cell extracts (100 µg of protein) were subjected to Western blot analysis with anti-DNMT1, anti-HA (to detect ectopic PKCδ), anti-PKCϵ, and anti-GAPDH antibodies. HCT116 cells were transfected twice at 24-hour intervals with 100 nM siRNA using Lipofectamine 2000 (Invitrogen) as described previously. 46 After 24 hours, cells were treated with 5-AzadC for 10 hours, and the cell extracts were subjected to Western blot analysis with anti-PKCδ and anti-DNMT1 antibodies.
Immunofluorescence analysis
H293T cells cultured directly on coverslips (50%-60% confluency) in a 6-well plate were transfected with pcDnmt1–N-118aa–Flag using Lipofectamine 2000 (Invitrogen). Immunofluorescence assay was performed as described previously.45,46
Co-immunoprecipitation assay
HCT116 cell extract was prepared in IPH buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA, 0.5% NP40, 1 mM PMSF) as described previously.45,47 In each case, 500 µL (500 µg of protein) of cell extract was precleared with protein G–agarose beads (GE Healthcare, Little Chalfont, UK) and subjected to immunoprecipitation overnight at 4°C using anti-DNMT1 or anti-PKCδ antibodies. Parallel control was run with IgG. Protein G–agarose beads were added to the reaction, and incubation was continued for 2 hours at 4°C. The beads were washed with IPH buffer; bound proteins were separated by SDS-PAGE and subjected to Western blot analysis with respected antibodies.
Isolation of nucleosomal protein and DNA
Nucleosomes were isolated as described previously 13 with slight modification. HCT116 cells (control and treated with 1 µM decitabine for 12 hours) were washed with PBS, suspended in ice-cold RSB buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2) supplemented with protease inhibitor cocktail (Sigma), and homogenized in a dounce homogenizer with 0.5% NP40. The nuclear pellet obtained after centrifugation was washed with the same buffer, and nuclei were resuspended in the RSB buffer supplemented with 0.25 M sucrose, 3 mM CaCl2, and 1 mM PMSF at 1 × 108/mL. Partial micrococcal nuclease digestion was performed by incubating with 10 units of enzyme at 37°C for 15 minutes, after which the reaction was stopped by adding EDTA/EGTA (10 mM). Nuclei were collected by centrifugation (1000g, 5 minutes) and suspended in 10 mM Tris-HCl (pH 7.4; containing 10 mM NaCl, 5 mM EDTA/EGTA), and nucleosomes were extracted by rocking at 4°C for 1 hour followed by centrifugation. Nucleosomes were then fractionated by layering onto 10 mM Tris-HCl buffer (pH 7.4) containing 0.25 mM EDTA, 300 mM NaCl, 5% to 30% sucrose, and followed by centrifugation at 39,000 rpm for 20 hours (40Ti Ultracentrifuge, Beckman Coulter, Brea, CA). Fractions were collected from the bottom of the centrifuge tube, and an equal amount of each fraction (a total of 18 fractions) was subjected to Western blotting. DNA was purified from each fraction by phenol-chloroform extraction and ethanol precipitation and separated by agarose gel electrophoresis.
Immunoprecipitation and in vitro kinase assay
H293T cells were transiently transfected with HA-tagged PKCδ expression vector plasmids or empty vectors as described previously.45,47 Forty-eight hours after transfection, cell extracts were prepared in IPK lysis buffer (20 mM Tris/HCl [pH 7.5], 150 mM NaCl, 25 mM β-glycerophosphate, 2 mM EDTA, 2 mM pyrophosphate, 1 mM orthovanadate, 1% Triton X-100, 1 mM DTT, 1 mM NaF; protease inhibitor cocktail). Protein G–agarose beads pre-equilibrated in the IPK lysis buffer were incubated with anti-HA antibody at 4°C for 1 hour. HA antibody–bound protein G–agarose beads were washed 3 times with the IPK lysis buffer and incubated in the same buffer with equal amounts (250 µg) of protein at 4°C for 3 hours. Proteins bound to the beads were washed 3 times with IPK lysis buffer and resuspended with 50 µM ATP, 1 µCi (γ-32P) ATP in kinase assay buffer (25 mM Tris-HCl [pH 7.5], 5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2), and 100 ng of recombinant DNMT1 protein as a substrate (New England BioLabs, Ipswich, MA) in a total volume of 50 µL for 30 minutes at room temperature. The supernatants were applied on P81 phosphocellulose paper squares (Millipore, Billerica, MA) and washed 3 times with 0.75% phosphoric acid followed by a wash with acetone and dried. Finally, the paper squares were suspended in scintillation cocktail, and the phosphate incorporation was measured in a scintillation counter.
Footnotes
Acknowledgements
We thank the late Dr. I. Bernard Weinstein (Columbia University) for providing the PKCδ expression vector plasmid. We also thank Drs. Masaki Okano (RIKEN) and Paula Vertino (Emory University) for generously providing mouse ES cells (WT and CS mutant of Dnmt1) and human fibroblast cells (HMT19 that overexpress DNMT1o), respectively. We also thank Huban Kutay and Satavisha Roy for technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported, in part, by grants CA101956 and CA86978.