
Abstract
Cancer cells exhibit altered metabolism characterized by the generation of adenosine triphosphate by glycolysis and generation of fatty acids by de novo synthesis. The majority of genes involved in these pathways have binding sites for specificity protein (Sp) transcription factors in their promoters. Studies showing that Sp transcription factors, particularly Sp1, are involved in the regulation in cancer cells of hexokinase, pyruvate kinase, lactate dehydrogenase, fatty acid synthase, and hypoxia-inducible factor-1α are reviewed. Glycolysis and lipogenesis in cancers are also known to be stimulated by the constitutive activation of the PI3K/Akt signaling pathway. Evidence is presented for the notion that Sp transcription factors may act in concert with Akt to regulate the abnormal metabolism of cancer cells.
There has recently been a resurgence of interest in the metabolism of cancer cells.1,2 A mechanistic understanding of the regulation of these metabolic pathways may lead to new strategies for cancer therapy and/or prevention. Otto Warburg first reported in the 1920s that cancer cells display altered metabolism. 3 He showed that most cancer cells, unlike normal cells that derive their adenosine triphosphate (ATP) from mitochondrial oxidative phosphorylation, generate the majority of their ATP by glycolysis, regardless of the availability of oxygen. Thus, cancers exhibit elevated uptake of glucose and produce large amounts of lactate. More recently, it has been shown that unlike nonlipogenic normal cells that preferentially derive fatty acids from the diet via the circulation, cancer cells derive their supply of fatty acids from de novo synthesis. 4 Thus, nontransformed cells express only low levels of fatty acid synthase (FAS), whereas most cancer cells express high levels of this enzyme.
Specificity protein 1 (Sp1), one of the first eukaryotic transactivators to be identified, is now known to be a member of the large multigene family of Sp/Kruppel-like factor (KLF) transcription factors (at least 20 in mammals).5,6 These proteins share a highly conserved DNA binding domain. Through 3 adjacent C2H2-type zinc fingers at the C-terminus, they bind to GC boxes, CACCC boxes (also called GT boxes), and basic transcription elements, collectively known as “‘Sp1 sites.” Sp/KLF proteins are subject to a number of posttranslational modifications including phosphorylation, glycosylation, and acetylation, enabling fine tuning of the regulation of gene transcription. Sp1 through Sp4 form a subgroup that contains glutamine-rich transactivating domains (TADs). Sp1, Sp3, and Sp4 have 2 TADs, whereas Sp2 has only 1 such domain and exhibits different DNA binding specificity. Sp5-9 are structurally similar but lack N-terminal glutamine-rich TADs. Sp1 and Sp3 are expressed ubiquitously, whereas Sp4 is expressed primarily in neural cells. Numerous mammalian genes are regulated by Sp proteins, often in cooperation with other transcription factors, and Sp proteins play important roles in a variety of physiological processes including cell cycle regulation and growth control, hormonal activation, apoptosis, and angiogenesis.5,6 Sp1 generally activates gene transcription, whereas Sp3 has both transcriptional repressor and activating properties. These activities depend on the promoter context, the cellular background, epigenetic factors, and interactions with other nuclear proteins.7,8 A variety of cancers have been shown to overexpress Sp proteins, particularly Sp1 and to a lesser extent Sp3 and Sp4. 7 Sp proteins are known to play a role in the regulation of multiple oncogenes and tumor suppressor genes as well as a number of cell cycle, apoptosis, and angiogenesis genes.5,7 Here, the evidence will be reviewed for a role for Sp transcription factors, primarily Sp1 and Sp3, possibly in conjunction with the constitutive activation of the PI3K/Akt signaling pathway, in regulating the abnormal glycolytic and lipogenic activity of cancer cells.
Glycolysis
The glycolytic pathway involves the metabolism of glucose to 2 molecules of lactate with a net gain of 2 molecules of ATP. The enzymes involved are illustrated in Fig. 1. How the metabolic requirements of proliferating cancer cells are fulfilled by glycolysis has been reviewed recently. 9 Since normal mitochondrial respiration cannot occur without oxygen, tumor hypoxia will cause a shift to glycolysis. Tumor cells, however, carry out aerobic glycolysis and, indeed, switch to glycolysis before hypoxia develops. The reactions catalyzed by hexokinase, phosphofructokinase, and pyruvate kinase are the major sites of regulation of glycolysis. These 3 reactions are exergonic and physiologically irreversible.
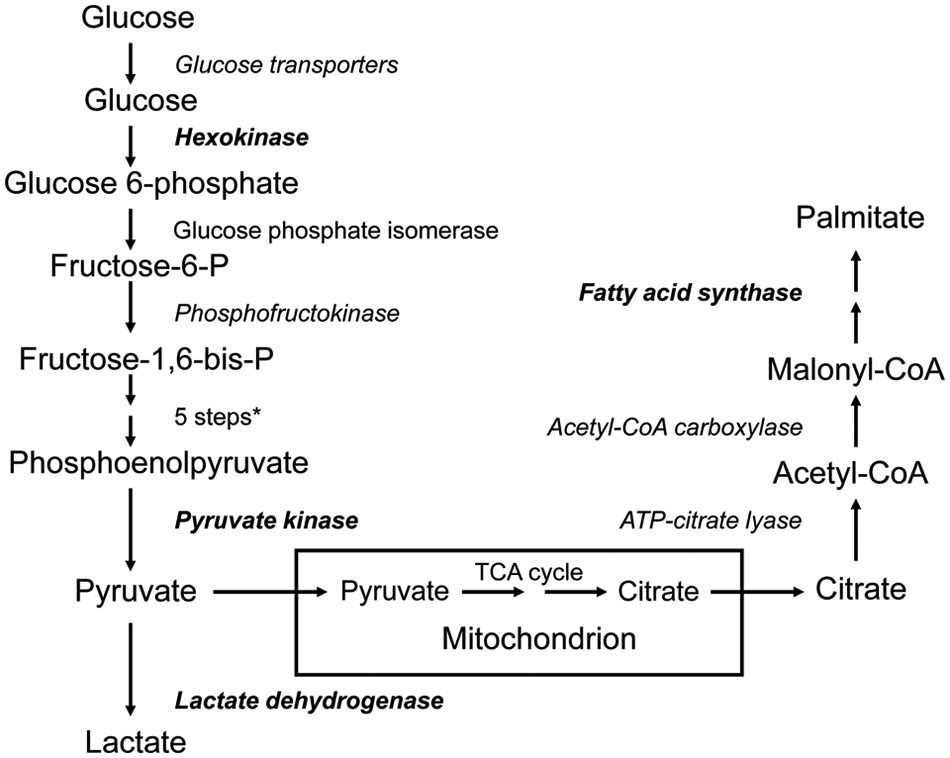
Overview of glycolysis and lipogenesis pathways. Genes known to be regulated by Sp transcription factors are shown in bold; proteins known to be aberrant in cancer cells are italicized. *Aldolase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, phosphoglycerate mutase, enolase. ATP = adenosine triphosphate; CoA = co-enzyme A; TCA = tricarboxylic acid.
Hexokinase
Of the 4 mammalian hexokinases, type II (HKII), which is bound to the outer mitochondrial membrane, is frequently expressed at levels more than 100-fold higher in rapidly growing tumors than in nontransformed cells.10,11 HKII is often the major isoform overexpressed and is required to maintain high levels of glycolysis. Indeed, the hyperactivity of HKII in tumors is the basis of positron emission tomography that is used for cancer detection. 10
Most of our knowledge of the regulation of HKII in cancer has come from studies in rat tumors. In highly glycolytic rat hepatomas, gene amplification 12 and promoter activation 13 have been shown to contribute significantly to HKII overexpression. Using rat hepatoma cells cultured in medium containing a serum supplement and glucose to simulate conditions in a well-vascularized tumor or within the peritoneal cavity, Lee and Pedersen, 10 using reporter gene assays, showed that a short segment of the promoter (−281 to −35 of the proximal region) contributes most to activation of HKII. They showed that 4 GC boxes, a CCAAT box, an inverted CCAAT box, and a cAMP-response element (CRE) are involved in promoter activation. Electrophoretic mobility shift assays (EMSAs) demonstrated binding of Sp1, Sp2, and Sp3 to 2 of the GC boxes and binding of Sp1 and Sp2 to the other 2 boxes. In addition, Lee and Pedersen 10 showed that NF-Y bound to the CCAAT box, and cAMP-response element-binding protein (CREB), activating transcription factor-1 (ATF1), and cAMP-response element modulator (CREM) bound to the CRE. Transfection studies showed that Sp1, Sp2, Sp3, CREB, and NF-Y are promoter activators that probably contribute to HKII overexpression in cancers.
Phosphofructokinase
6-phosphofructo-1-kinase (PFK-1) is the major rate-limiting step of glycolysis. Three isoforms of mammalian PFK-1 encoded by different genes have been identified P (high levels in brain), M (only form in muscle), and L (major form in liver). PFK-1 activity is controlled by the intracellular ATP/AMP ratio—high levels of ATP inhibit PFK-1 activity. Fructose 2,6-bisphosphate (F2,6BP) is a powerful allosteric regulator that acts by increasing the affinity of PFK-1 for fructose 6-phosphate and decreasing the inhibitory effect of ATP. 14 The bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase) determines the steady-state concentration of F2,6BP. PFK-2/FBPase is expressed in several tissue-specific isoforms encoded by at least 4 genes (PFKB1-4) that are all stimulated by hypoxia through hypoxia-response elements in their promoters. 15
PFK-1 is known to be expressed in human lymphomas and gliomas,16,17 and PFK-1 activity is higher in breast cancer metastases than in the primary tumors. 18 Expression of the PFK-1L isoform has been shown to correlate with the glycolytic activity of human breast cancer cell lines 19 and is preferentially expressed in gliomas. 17 A number of cancer cell lines produce elevated levels of F2,6BP compared with normal cells, suggesting that PFK-2/FBPase is activated or overexpressed. 20 PFKFB3 is known to be highly expressed and play an important role in the metabolic regulation of human cancers and cancer cell lines.21,22 Likewise, PFKFB4 is overexpressed in several tumors, including breast tumors. 23
There are no reports of the involvement of Sp transcription factors in the expression of PFK-1 in cancer. However, PFK-1P is highly abundant in brain, and the PKF-1P gene has been shown to contain 7 Sp binding sites in its promoter. 24 Sp1 and Sp3 were shown to bind to most of these sites, and deletion and mutation analysis suggested that they contribute positively to promoter activity. The existence of an Sp1 binding site in a promoter, however, does not necessarily imply that an Sp family member is involved in the regulation of that gene. Indeed, the promoter of the human placental form of PFK-2/FBPase contains Sp1 binding sites that appear not to be essential for transcription of the gene. 25
Pyruvate Kinase
Pyruvate kinase (PK) is the final rate-limiting step of glycolysis. In mammals, there are 4 PK isoforms (L, R, M1, and M2) encoded by 2 genes, PKL encoding the L and R isoforms and PKM encoding the M1 and M2 isoforms. PKL and PKR are mainly expressed in liver and red blood cells. PKM1 is expressed mainly in muscle and brain, whereas PKM2 is expressed during embryonic development. 26 Both the rat and human genes contain 12 exons and 11 introns, exons 9 and 10 containing sequences specific to the M1 and M2 isoforms, respectively. 27 It appears, therefore, that tissue-specific, mutually exclusive, alternative splicing selects exons 9 and 10 to produce the 2 isoforms from a common primary transcript. 28 In contrast to differentiated cells, proliferating tumor cells express exclusively PKM2. 26 Indeed, it has recently been shown that switching PKM2 expression to PKM1 in tumor cells reverses the Warburg effect and reduces the ability of the cells to form tumors in nude mice, indicating that PKM2 expression is necessary for aerobic glycolysis and maximal tumor growth. 26 It has also recently been shown that tyrosine kinase signaling regulates PKM2 activity. 29
The 5′-flanking region of the human PKM gene contains putative Sp1 binding sites. 27 A series of studies in rat thymocytes 30 showed that the PKM promoter has 5 Sp binding sites, 3 of which were functional in transfection assays and were stimulated by Sp1 and Sp3. In rat hepatoma cells that have high glycolytic activity, glucose treatment caused increased binding of Sp1 to its consensus sequence because of its dephosphorylation, resulting in the transcriptional activation of PKM. 31 The same mechanism of transcriptional activation was also reported for aldolase A. Disher et al 32 showed that transient transcription of a reporter gene directed by the PKM promoter was activated when transfected myocytes were exposed to hypoxia. The promoter, however, does not contain a hypoxia-inducible factor-1 binding site, and the hypoxia response was localized to a conserved GC-rich element that bound Sp1 and Sp3. Hypoxia caused depletion of Sp3, whereas Sp1 levels remained unchanged, suggesting that hypoxia activates PKM by down-regulating Sp3, thereby removing its transcriptional repression.
Other Genes of the Glycolytic Pathway
A number of other genes in the glycolytic pathway have been shown to have Sp1 sites in their promoters. including human glucose phosphate isomerase, 33 rat and human aldolase A,30,31,34 and aldolase C35,36 and the following human genes: phosphoglycerate kinase-1 37 and phosphoglycerate kinase-2, 38 α-enolase, 39 β-enolase, 32 and enolase-3. 40 The actual involvement of Sp1 in the regulation of these genes, however, appears not to have been investigated. Furthermore, a number of them are expressed in a tissue-specific manner. Recently, the human testis-specific lactate dehydrogenase c gene (hLdhc), which is highly expressed in human lung cancer, melanoma and breast cancer, 41 was shown to be regulated in cancer cells by Sp1 as well as by a CRE and CpG island methylation. 42
Lipogenesis
Cellular synthesis of long chain fatty acids involves the rate-limiting conversion of acetyl–co-enzyme A (CoA) into malonyl-CoA by the enzyme acetyl-CoA carboxylase (ACC), followed by the FAS-catalyzed condensation of 1 mole of acetyl-CoA and 7 moles of malonyl-CoA to form 1 mole of palmitate; ATP citrate lyase (ACL) catalyzes the conversion of citrate to acetyl-CoA, thereby linking glycolysis and lipogenesis (Fig. 1).
Fatty Acid Synthase
FAS is down-regulated in most normal human tissues by the uptake of circulating fatty acids from the diet. Highly proliferating cancer cells derive their fatty acids for membrane production and posttranslational modification of proteins from elevated, deregulated de novo synthesis. 43 Inhibition of FAS suppresses cell proliferation and tumor growth in xenograft models of breast and ovarian cancers and pleural mesothelioma44-46 and suppresses growth and induces apoptosis in various cancer cell lines. 43 Overexpression of FAS has been shown in numerous cancers, including those of the prostate, ovary, colon, lung, endometrium, and stomach. 4 Furthermore, poor prognosis is often associated with elevated FAS expression. 4
Control of expression of the FAS gene in lipogenic tissues such as liver and adipose tissue by dietary and hormonal signals is accomplished via several transcription factors that are known to bind to defined regions within the FAS promoter. These include SREBP-1c, Sp1, and NF-Y.47,48 SREBP-1c is a relatively weak transcriptional activator but functions efficiently in concert with NF-Y and Sp1. 47
A recent report suggests that Sp1 plays a role in regulating both proliferation and de novo lipogenesis in cancer cells. 49 In MCF-7 human breast cancer cells transfected with Sp1 small interfering (siRNA), decreased Sp1 levels were associated with a decrease in growth rate and CDC25A expression with an inhibition of G1/S transition and S phase progression. FAS expression was also significantly suppressed in the transfected cells. Although Sp3 and Sp4 were shown to inhibit proliferation and CDC25A expression in MCF-7 cells, FAS expression was not affected. Treatment of MCF-7 cells with mithramycin, a compound that is known to block GC-rich promoter regions and suppress Sp1 activity, 50 decreased proliferation and inhibited CDC25A and FAS expression. Using chromatin immunoprecipitation (ChIP) assays, the investigators showed that Sp1 binding to the promoters of both CDC25A and FAS genes was significantly inhibited by mithramycin.
Many reports show that SREBP-1c regulates FAS expression in normal cells 47 as well as cancer cells. 51 Deng et al, 52 however, showed that insulin-stimulated SREBP-1c expression in rat hepatocytes is mediated by Sp1. In MCF-7 cells, transfection with Sp1 siRNA or treatment with mithramycin suppressed SREBP-1c expression, suggesting that Sp1 regulates FAS by a dual mechanism involving SREBP-1c and direct binding. 49 Neither Sp3 nor Sp4 siRNAs affected SREBP-1c.
It is well documented that Sp1 mediates gene expression in response to various hormones. 53 Lu and Archer 49 showed that MCF-7 cells treated with estradiol increased their expression of CDC25A and FAS with increased binding of Sp1 to the promoters of the 2 genes, without any increase in the expression of Sp1. Swinnen et al 54 showed that androgens stimulate the expression and activity of FAS in LNCaP cells via the androgen receptor (AR). Since the AR and Sp1 can complex with each other, recruiting co-activators and general transcription factors, 55 it seems likely that androgen stimulation of FAS involves Sp1.
In an extension of their studies, Lu and Archer 49 investigated whether Sp1 plays a role in regulating FAS expression and proliferation in colon and prostate cancer cells. Treatment of colon cells with Sp1 siRNA inhibited proliferation as well as FAS and SREBP-1c expression but not CDC25A expression. In prostate cells, Sp1 siRNA treatment suppressed proliferation and FAS and CDC25A expression but not SREBP-1c expression. These results indicate cell type specificity in the actions of Sp1. Overall, the studies of Lu and Archer suggest that Sp1 may coordinately regulate fatty acid synthesis and proliferation in cancer cells.
Another report links FAS and Sp1, albeit somewhat indirectly, in cancer cells. Choi et al 56 showed that the transcription factor/proto-oncogene FBI-1 (Pokemon/ZBTB7A) and SREBP-1 synergistically activate transcription of FAS. Electrophoretic mobility shift and ChIP assays showed that SREBP-1, Sp1, and FBI-1 bind to the GC and SRE boxes of the FAS promoter. Binding competition among the 3 transcription factors appears to be important in the transcriptional regulation.
ATP Citrate Lyase and Acetyl-CoA Carboxylase
In addition to elevated FAS, elevated ACC and ACL expression and activity have been reported in cancer cells. 57 Sp1 sites are known to be present in the promoters of these genes.58-60 In Drosophila SL2 cells, Sp1 and Sp3 together with SREBP-1 transcriptionally activate ACC and FAS promoters. 61 Glucose induces an increase in Sp1 binding to ACC in adipocytes by dephosphorylation of nuclear Sp1. 59 In human hepatoma cells, Sp1 and Sp3 have been shown to play an important role in the regulation of ACL by glucose. 58
Other Genes Involved in Regulating Cancer Cell Metabolism
Glucose Transporters
Although not strictly part of the classical glycolytic pathway, glucose transport in the tumor cell is clearly necessary for glycolysis to take place. Specific glucose transporters mediate glucose entry into most glucose-sensitive tissues. In a wide variety of human tumor samples examined, GLUT1, 2, and 5 (fructose transporter) were the main transporters detected in 58%, 31%, and 27% of samples, respectively.62,63 GLUT3 and GLUT12 are overexpressed in some tumors. 62 Transcriptional regulation of the GLUT isoforms involved in cancers has not been studied. However, the rat GLUT1 and GLUT2 promoters contain Sp1 sites that may be responsible for regulation of basal transcription rates in a number of tissues.64,65 It has also been shown that Sp1 and Sp3 bind to the mouse GLUT3 gene, Sp1 mediating suppression and Sp3 mediating activation of expression of this gene in murine neuroblasts and trophoblasts. 66
Hypoxia-Inducible Factor-1α (HIF-1α)
HIF-1α is a transcription factor that is an important regulator of the response of tumors to hypoxia, including resistance to apoptosis and increased angiogenesis. Importantly, HIF-1α increases the expression of a number of genes that encode glycolytic enzymes 67 as well as GLUT1, 68 and in human breast cancer cell lines, the FAS gene is up-regulated by phosphorylation of Akt followed by activation of HIF-1α then induction of SREBP-1. 69
The HIF-1α promoter contains binding sites for several transcription factors, including Sp1. 70 Koshikawa et al 71 showed that the constitutive up-regulation of HIF-1α in highly metastatic mouse Lewis lung carcinoma cells is abolished by mithramycin, although luciferase reporter and ChIP assays showed that Sp1 was necessary but not sufficient for HIF-1α mRNA overexpression. In support of these observations, Kim and Park 72 recently showed that in PC3 human prostate cancer cells, HIF-1α transcription is dependent on Sp1. Thus, HIF-1α transcription was suppressed in cells treated with mithramycin and enhanced HIF-1α transcription was observed in cells treated with trichostatin A, which induces Sp1 activation. 73 Kim and Park also found that siRNA knockdown of mitochondrial NADP+-dependent isocitrate dehydrogenase (IDPm) suppressed hypoxia-induced stimulation of HIF-1α in PC3 cells. In cells transfected with an Sp1-responsive luciferase reporter gene, knockdown of IDPm inhibited Sp1-mediated luciferase activity, providing further evidence that HIF-1α transcription is dependent on Sp1.
Altered Cancer Cell Metabolism May Be Mediated by Both Sp Proteins and Akt
Sp proteins are regulated by, or interact with, a number of oncogenes and tumor suppressor genes that play a role in controlling the metabolic switch caused by malignant transformation.5,6,74 More specifically, glycolysis and lipogenesis are known to be stimulated by the constitutive activation of the PI3K/Akt signaling pathway that is present in a large proportion of human cancers.75,76 Indeed, Akt has been shown to be sufficient to stimulate the switch to aerobic glycolysis in cancer cells, and their survival and continued growth depend on aerobic glycolysis induced by Akt. 77 Likewise, Akt signaling modulates FAS expression in cancer cells, at least in part via activation of SREBP-1 78 and possibly in concert with Sp1 (vide supra). Indeed, constitutive activation of the Akt pathway protects against cell death induced by FAS inhibitors, 79 and, conversely, inhibition of Akt sensitizes cancer cells to apoptosis induced by inhibitors of FAS. 80 Overexpression of FAS in prostate cancer has been shown to be linked to the phosphorylation and nuclear accumulation of Akt. 81
The evidence for a role for both Sp1 and Akt in regulating the altered metabolism of cancer cells suggests they may be mechanistically linked. This notion is supported by a relatively large number of examples in which Sp1 is required for the transactivation of various genes in cancer cells by the PI3K/Akt pathway. Thus, Mireuta et al 82 recently showed that IGFBP-2 expression in MCF-7 human breast cancer cells is regulated by PI3K/Akt through an Sp1-induced increase in transcription. Other examples of an Sp1-Akt link include the regulation of β1,4-galactosyltransferase in gliomas, 83 membrane-type-1 matrix metalloproteinase (MMP-1) in prostate cancer cells, 84 MMP-2 in renal clear cell carcinoma cells, 85 HIF-1α in Lewis lung carcinoma cells, 71 and the stress-response gene Redd 1 in HeLa cells. 86 It seems likely, therefore, that the alterations in cell metabolism caused by malignant transformation are mediated, at least in part, by both Sp1 and Akt.
Conclusions
The majority of genes in cancer cells that are involved in the generation of ATP by glycolysis and the abnormal generation of fatty acids by de novo synthesis have binding sites for Sp transcription factors in their promoters, and there is evidence that these transcription factors are involved in the regulation of a number of metabolic genes in cancers, including hexokinase, pyruvate kinase, lactate dehydrogenase, fatty acid synthase, and hypoxia-inducible factor-1α. This suggests that Sp proteins play an important role in regulating cancer cell metabolism. Glycolysis and lipogenesis in cancer cells are known to be stimulated by the constitutive activation of PI3K/Akt signaling, and there is evidence for the notion that Sp transcription factors, particularly Sp1, may act in concert with Akt to regulate the alterations in cell metabolism caused by malignant transformation. Increased understanding of the mechanism by which Sp transcription factors, together with Akt, are involved in the regulation of human cancer cell metabolism may lead to novel therapeutic approaches to control the disease.
Footnotes
Acknowledgements
I thank Drs Robin Duncan and Suying Lu for helpful advice.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.