
Abstract
Covalent modifications of histones can regulate all DNA-dependent processes. In the last few years, it has become more and more evident that histone modifications are key players in the regulation of chromatin states and dynamics as well as in gene expression. Therefore, histone modifications and the enzymatic machineries that set them are crucial regulators that can control cellular proliferation, differentiation, plasticity, and malignancy processes. This review discusses the biology and biochemistry of covalent histone posttranslational modifications (PTMs) and evaluates the dual role of their modifiers in cancer: as oncogenes that can initiate and amplify tumorigenesis or as tumor suppressors.
Introduction
Covalent posttranslational modifications of proteins are a major mechanism by which protein function is regulated.1-4 These modifications can, for example, facilitate protein interactions or determine protein stability and thereby regulate specific cellular responses. They can be highly dynamic or have a function in epigenetic memory as stable marks. These properties enable precise regulation of key processes by these modifications such as cell cycle, apoptosis, signal transduction, and chromatin structure.
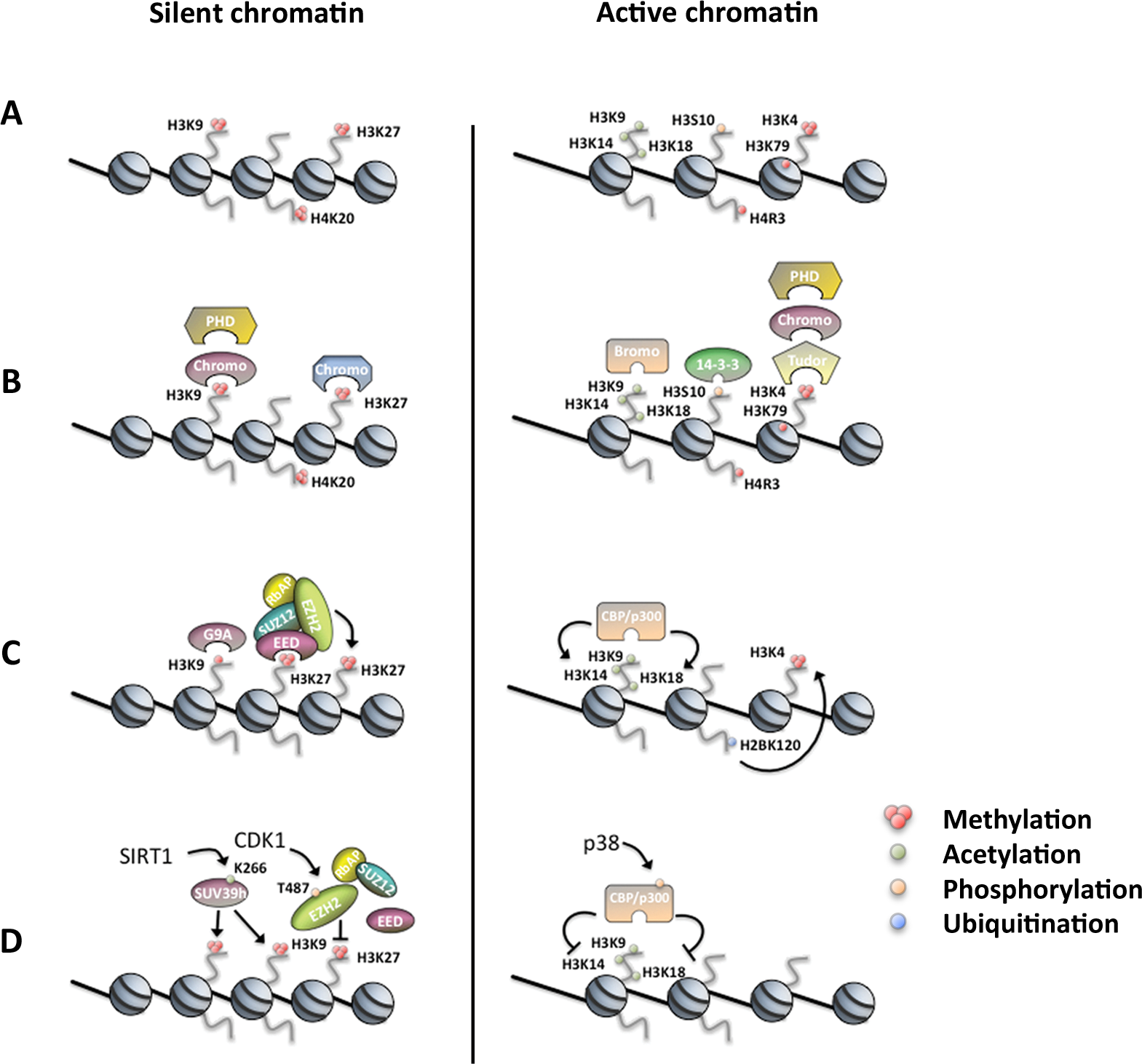
Currently, the best-characterized substrates for multisite protein modifications are the histone proteins. Although histones were originally considered simply as a static scaffold for DNA packaging, it is now becoming evident that they are dynamic proteins, which undergo multiple types of posttranslational modifications (PTMs) in order to play a fundamental role in regulating chromatin function. 5 Nucleosomes are the basic units that compose the chromatin fiber. Each of these primary building units consists of 4 core histones (H3, H4, H2A, and H2B), which form a histone octamer, assembled from 2 heterodimers of the core histones H3 and H4 together with 2 heterodimers of H2A and H2B, around which 146 base pairs of DNA are wrapped. 6 The unstructured N- and C-terminals of the histone tails extend from the DNA gyres and are heavily posttranslationally modified with several different modifications such as acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deimination, biotinylation, butyrylation, N-formylation, and proline isomerization (Fig. 1A). 4 These covalent modifications are added or removed by enzymes often termed as “writers” and “erasers.” By establishing different combinations of histone PTMs, they can form the “histone code.”1,3 In addition, recent reports also show a substantial impact of PTMs occurring on residues within the histone-structured globular domain.7,8 While histone tail modifications serve mainly as “signaling platforms” and determine the interactions of histones with other proteins (Fig. 1B), modifications in the globular domain may directly affect the DNA-binding surface of the nucleosomes by altering the physical interaction between the histone body and DNA. 9
Histone PTMs regulating chromatin-dependent processes. (
In recent years, chromatin research focused mainly on the core histones, whereas the linker histone H1, which binds to the nucleosome at the entry/exit point of DNA, has been largely overlooked. Similar to the core histones, H1 histones are targets of multiple posttranslational modifications, including phosphorylation, acetylation, methylation, ubiquitination, and ADP ribosylation. Analysis with modern mass spectrometry has revealed a large number of modified residues, both in the tails and the globular domain of H1.10,11 H1 modifications are likely to be involved in the regulation of the multiple linker histone functions 12 ; therefore, their characterization is necessary in order to better understand higher-order chromatin structure and dynamics.
The potential complexity of histone PTMs in regulating chromatin-dependent processes is reflected by more than 10 different types of modifications (listed above) at nearly 80 different sites that were reported on histone tails and the core domains (including the linker histone H1). 4 The number of histone PTMs is likely to rise in the following years because a huge effort is being made in searching for novel PTM sites, with significant improvement of several detection and genome-wide mapping techniques. 13
Because PTMs occur on distinct amino acid residues on specific histones and within chromatin at certain genomic regions, this allows a vast range of flexibility in regulating chromatin dynamics and signaling transmission. By employing different combinatorial patterns of modifications, it is possible to regulate two opposite processes like transcriptional activation and repression using the same amino acid sequence within a histone. For example, acetylation of key lysine residues of histone H3 and H4 by histone acetyltransferases (HATs) is commonly associated with transcriptional activation, whereas low levels of acetylation are found at transcriptionally inactive chromatin (Fig. 1A). 4 Another level of complexity and regulation can be found in the cross-talk between histone marks, where modifications regulate one another.2,14 A specific modification can directly influence the occurrence of another PTM either in cis, within the same histone, or in trans, between 2 adjacent histone molecules or even across different nucleosomes. 15 This additional regulatory layer adds a greater combinatorial intricacy and larger diversity to the histone language.
Additionally, posttranslationally modified chromatin can also serve as a selective binding platform for regulatory proteins, which can then translate this signal into changes in chromatin structure or dynamics. Evolutionarily conserved specialized proteins, termed “histone readers,” possess the ability to specifically bind certain histone modifications and mediate the initiation of a defined nuclear process such as transcription, DNA repair, or replication (Fig. 1B). 16 For example, it is now well established that trimethylated H3K9 (H3K9me3) can recruit and be bound by a protein known as heterochromatin protein 1 (HP1) via its evolutionarily conserved chromodomain.17,18 This interaction with H3K9me3 was shown to occur in “repressed” chromatin (i.e., heterochromatin) and leads to formation of compacted chromatin that can physically inhibit the access of the transcriptional machinery. In addition to H3K9me3, K26-methylated H1 can also be bound by HP1. 19 Interestingly, histone-modifying complexes that set or remove these marks were recently shown to possess specific “reader” capabilities. For example, binding of trimethylated H3K27 (H3K27me3) by the Polycomb repressive complex 2 (PRC2) stimulates the activity of the enzymatic PRC2 subunit enhancer of zeste homolog 2 (EZH2), which in turn places more H3K27me3 marks on the neighboring nucleosomes.20,21 These findings show that patterns of histone modifications can be “read and written” by the same interacting factor(s) or complexes (Fig. 1C).
Recently, several reports demonstrated regulation of the “writers” and “readers” themselves by PTMs, which adds an additional layer of regulation. For example, p300 that acts as a crucial element in the eukaryotic gene regulation network was shown to be regulated by phosphorylation, which greatly reduces its HAT activity.22,23 The mitogen-activated protein kinase p38 associates with p300 and is implicated in the phosphorylation-mediated degradation of p300 stability and function (Fig. 1D). 24 Also, EZH2 is now known to be phosphorylated by cyclin-dependent kinase 1 (CDK1) in a cell cycle–dependent manner (Fig. 1D).25,26 These sequential PTMs were shown to affect either EZH2 enzymatic activity or its recruitment to target genes by increasing the binding to noncoding RNA (also see below).
While it is still under discussion whether histone modifications form a true “code,” it has become clear that they control (almost) all chromatin-mediated processes such as gene expression, DNA repair, chromosome condensation, development, and differentiation as well as disease processes. Numerous reports show a tight link between age-related diseases, birth defects, and several types of cancer with disruption of certain histone PTMs or patterns of PTMs.27-30 Therefore, we believe that one of the major goals in postgenomic biology is to unravel the molecular basis and the physiological role of covalent protein modifications. Identifying novel histone modifications, the modifying enzymes, and the downstream effector proteins that can bind to these marks and translate them into specific biological outcomes sets a fascinating challenge. This has the potential to identify novel disease pathways and thereby new therapeutic targets as well as diagnostic markers.
In this review, we discuss the different types of covalent histone modifications (in particular, acetylation, phosphorylation, and lysine methylation) and the corresponding modifiers with a specific focus on their role in cancer. In addition to the modifications of histones, we also describe modifications of the modifiers that regulate their activity, adding another level of complexity to the language of PTMs.
Histone Acetylation and Acetyltransferases
Lysine acetylation is a reaction in which an acetyl group from the acetyl-coenzyme A (acetylCoA) cofactor is transferred to the e-amino nitrogen of lysine residues. This reaction is catalyzed by histone acetyltransferases (HATs), whereas the reverse reaction is performed by histone deacetylases (HDACs).
Already in the early 1960s, it has been observed that histones can be covalently modified by acetyl groups,31,32 and very soon, this modification has been linked to transcriptional activation. 33 However, the first HAT catalytic proteins were isolated and cloned only in the 1990s.34-36 The revolutionary finding that a transcriptional coactivator protein, Gcn5, is a nuclear HAT 35 led to a surge in HAT-related research and the identification of numerous enzymes and their substrates. HATs can be grouped on the basis of their catalytic domains into 3 families: Gcn5 N-acetyltransferases or GNATs (including Gcn5, PCAF, Hat1, and others); MYST HATs (named for the founding members of this family: MOZ/Morf, Ybf2, Sas2, and Tip60); and an “orphan class” of HAT enzymes lacking a true consensus HAT domain, featuring, among others, p300/CBP and Taf1. 37 Representative members of the families, with important physiological roles as well as putative links to carcinogenesis, are subject to a more detailed discussion below. In general, opposite to HATs, HDACs negatively regulate gene expression. Four HDAC classes have been identified: class I HDACs (HDAC1-3 and 8) are related to yeast Rpd3, class II HDACs (HDAC4-7, 9, and 10) are related to yeast Hda1p, and class IV HDACs contain one unrelated member (HDAC11). The enzymes in those classes are zinc dependent. Class III HDACs (sirtuins) are NAD-dependent enzymes related to yeast Sir2. 38 Sirtuins and their involvement in cancer are the subject of another review in this issue (see Bosch-Presegué & Vaquero).
A common feature of HATs and HDACs is that they operate within multiprotein complexes and often the same enzyme resides within more than one complex. The complex in which a given HAT or HDAC is incorporated determines in many cases its substrate specificity and/or its interaction with other protein factors—and as such has a profound effect on its function.37,39 In general, HATs, and to an even greater extent, HDACs, are promiscuous enzymes when compared to the highly specific histone methyltransferases (HMTs).
HATs and HDACs were initially found to target histones as substrates but subsequently were also shown to target many nonhistone proteins, a fact that eventually led to a change in their nomenclature and renaming as lysine acetyltransferases and lysine deacetylases (the new nomenclature is not commonly employed in the field yet; therefore, in this review, we use the traditional nomenclature). 40
Apart from the core histones, linker histone H1 can also be acetylated. The first demonstration of H1 lysine acetylation in vivo came for H1.4K26. 41 In this study, it has been shown that SirT1, a HDAC involved in establishing repressive chromatin, interacts with H1.4 and is able to deacetylate H1.4K26 in vitro. However, it is still unknown which enzyme acetylates H1.4K26 and what is the biological function of this mark. A recent mass spectrometry study revealed that there are many acetylation sites on H1, 11 awaiting functional studies.
There is now a wealth of evidence pointing to a role of core histone acetylation in transcriptional activation, and two molecular mechanisms have been suggested. On the one hand, acetylation partially neutralizes the positive charge of histones, weakening their interaction with the nucleosomal DNA, thus probably “opening” chromatin structure and facilitating the access of transcription factors to their recognition elements. 42 On the other hand, acetylated lysines can create a specific signal for regulatory factors or chromatin-remodeling complexes and contribute to their targeting to a specific region. Consistent with this hypothesis, it has been found that bromodomains act as acetyl-lysine recognition modules, directing enzymes that contain them to particular chromosomal sites.43,44 Besides transcription, new roles for histone acetylation have been uncovered and include nucleosome assembly, chromatin folding, heterochromatic silencing, 39 DNA damage repair, and replication.37,45,46
We now move from general functions of histone acetylation to a discussion of representative HAT enzymes, together with their substrates, and highlight their potential role in oncogenesis.
Gcn5 and the SAGA Complex
The Tetrahymena thermophila protein p55 was the first identified nuclear, transcription-related HAT enzyme and shown to be the homolog of yeast Gcn5 (general control nonderepressible 5). 35 An intriguing observation at that time was the inability of recombinant Gcn5 to acetylate nucleosomal histones, while it exhibited strong HAT activity on recombinant histones. It has been subsequently shown that native Gcn5 exists in high molecular weight complexes, whose subunits enable Gcn5 to acetylate nucleosomes. 47 The more notable complex is SAGA (named for Spt-Ada-Gcn5 acetyltransferase), a 2-MDa multiprotein, chromatin-modifying molecular machine, highly conserved between yeast and humans. Human SAGA was also named in the literature as TFTC (TATA-binding protein-free TAFII-containing complex).48-50 A second distinct Gcn5-containing complex in metazoans is ATAC (Ada2a containing). 51
While most metazoan genomes code for one Gcn5-type factor, vertebrates have a second gene encoding pCAF (p300/CBP-associated factor), which has 73% identity to Gcn5. 50 Gcn5 is an essential protein both in Drosophila 52 and mice 53 ; moreover, it is able to compensate for pCAF loss in mammals: pCAF−/− mice have elevated Gcn5 levels, allowing them to develop normally. 53
The substrate specificity of Gcn5 in vitro depends on the complex it is in and, to a lesser extent, on the organism from which it is purified. As part of the SAGA complex, Gcn5 acetylates preferentially 4 lysine residues in the N-terminal tail of histone H3: H3K9, K14, K18, and K23. 50 In vivo, Drosophila Gcn5 was found to be involved in acetylation of larval polytene chromosomes at H3K9 and H3K14, and surprisingly also H4K5 and H4K12, which was not predicted from biochemical studies.52,54 In chicken cells, Gcn5 loss leads to decreased acetylation levels of H3K9 and H2BK16. 55 Disruption of Gcn5 HAT activity in mice leads to decreased acetylation levels of H3K9/K18 and H4K12, but H3K14 remains unchanged. 56 In human cells, Gcn5 is involved in the acetylation of H3K9 and K14, both in the ATAC and SAGA complexes. 57 In addition to being a “writer” of the histone code, SAGA also contains multiple domains capable of recognizing acetylated and methylated histone PTMs so it can “read” the combinatorial histone code.48,58
Apart from the well-established functions of Gcn5 and the SAGA complex in core histone acetylation and transcriptional activation, new roles are emerging for this complex. For example, while SAGA is mostly known for activating highly regulated genes that respond to environmental stresses59,60 and is required for transactivation of nuclear receptors,61,62 it can also play an inhibitory role in the transcription of some inducible genes, such as HIS3 and ARG1.63,64 In particular, while Gcn5 is required for normal HIS3 transcriptional start site selection and transcriptional activation, another SAGA subunit, Spt8, inhibits TBP binding to the HIS3 promoter. 63 Furthermore, SAGA is now emerging as a regulator of a whole set of mRNA-related processes, including transcription elongation, mRNA splicing, and export.65-67 Finally, Gcn5 and SAGA appear to be important for the maintenance of genome integrity through promoting replication-coupled nucleosome assembly, assisting in DNA repair especially after UV damage, as well as participating in telomere maintenance.68-70
Taking into account the SAGA complex’s functional importance, it is not surprising that mutations or altered expression of SAGA subunits correlate with neurological disease and aggressive cancers in humans.49,71 SAGA functions as a cofactor for several proto-oncoproteins. 50 In particular, the subunits TRRAP and Gcn5 regulate the oncogenic activity of the oncoprotein c-Myc and tumor suppressor p53, 72 and another SAGA subunit, SPT7, is implicated in the transcription of several Myc-dependent genes. 73 BRCA1 is a tumor suppressor protein with E3 ubiquitin ligase activity and is involved among others in DNA repair and cell cycle progression. Mutations in the C-terminal transactivation domain of BRCA1, which predispose women to early onset of breast cancer, result in a loss of interaction between BRCA1 and TRRAP, which in turn prevents Gcn5/TRRAP from coactivating BRCA1. In fact, it seems that the HAT activity of Gcn5 is necessary for BRCA1-mediated gene regulation and DNA repair. 74 Further on, the deubiquitinase subunit of SAGA, USP22, has been grouped by transcriptional profiling into an 11-gene signature of poor prognosis in multiple types of cancer.75,76 USP22, together with Gcn5, protects telomeres from DNA damage through the stabilization of the TRF1 component of shelterin, which is dependent on USP22-mediated deubiquitination of TRF1. 77
The Histone Acetyltransferase MOF
MOF (males absent on the first) is a member of the MYST family of HATs and has very particular characteristics. The Drosophila MOF protein is particularly well studied as a key component of the dosage compensation complex, which is required to upregulate X-linked gene expression in male flies having one X chromosome, in contrast to females that have two. 78 While mammals use a different mechanism for dosage compensation (one of the two X chromosomes gets inactivated in females), the MOF enzyme is conserved between humans and flies, together with the multiprotein complexes in which it resides.79-81
An additional striking feature of MOF is its high acetyltransferase specificity for lysine 16 on histone H4 (H4K16), a unique and unusual characteristic for a HAT enzyme.78,82 Moreover, MOF appears to be the main acetyltransferase for H4K16, as MOF knockout mice embryos exhibit a loss of acetylation specifically at this residue. 83 Nota bene, those MOF-null embryos die at implantation in vivo; hence, MOF is essential for mammalian development. The acetylation of H4K16 has profound effects on chromatin structure: it inhibits in vitro the formation of compact 30-nm-like fibers and impedes the ability of chromatin to form cross-fiber interactions. 84 Interestingly, acetylation of only 30% of H4K16 disrupts nucleosomal array folding to the same extent as the complete removal of all N-terminal histone tails and inhibits chromatin compaction to a greater extent than deletion of the H4 N-terminal tail altogether. 85 By now, no other single histone acetylation or methylation has been found to affect so profoundly the chromatin structure as H4K16ac.
MOF is involved in multiple cellular processes. Most notably, H4K16 acetylation mediated by MOF leads to increased transcription both in vitro and in vivo.81,82 However, apart from transcription, MOF participates in DNA damage response and, together with another MYST family HAT, Tip60, acetylates p53 on K120 to influence the cell’s decision to undergo apoptosis versus cell cycle arrest.86-89 One could hypothesize that deregulation of this type of processes might influence cancer progression. Concomitantly with this, the loss of acetylation at H4K16 (as well as H4K20 methylation) has been found to be a common hallmark of human cancer. 90 This reduction occurs at hypomethylated DNA repetitive regions, which have been previously linked to chromosomal instabilities.91,92 It is currently not clear whether and how MOF is deregulated in those cancers; mutations in MOF, downregulation of its enzymatic activity, or its mistargeting could all account for H4K16ac loss. For comprehensive reviews on MOF and other MYST family members’ involvement in cancer, see Avvakumov and Côté 93 and Rea et al. 94
The Histone Acetyltransferases p300 and CBP
p300 and CBP were originally identified as binding partners of the adenovirus early region 1A (E1A) protein95,96 and the cyclic AMP response element (CRE)–binding protein, 97 respectively. They were then shown to function as transcriptional adaptors, and in 1996, 2 independent groups have demonstrated that p300 and CBP possess HAT activity.98,99
p300 and CBP have a multidomain structure and contain, in addition to the enzymatic HAT domain, several protein interaction domains. 100 p300 and CBP are the most promiscuous HAT enzymes; they are capable of acetylating all the 4 core histones on multiple residues 101 as well as at least 70 other proteins, including p53, Rb, E2F, and myb. 102 Most of p300 and CBP target sites on histones can be also acetylated by other enzymes, and a recent study has identified H3K18ac and H3K27ac as the only histone acetylation sites significantly reduced upon CBP and p300 depletion in mouse embryonic fibroblasts. 103 p300 and CBP are highly homologous, in particular their HAT domains, which share >90% sequence identity, and are often referred to as p300/CBP, indicating a presumable functional redundancy. However, in vivo studies have revealed that CBP and p300 have both shared as well as unique properties. For example, while both CBP and p300 knockout mice die before birth, displaying an open neural tube defect,104,105 only p300−/− (and not CBP−/−) embryos exhibit defective development of the heart. In contrast, CBP+/− mice display growth retardation and craniofacial abnormalities as well as an increased incidence of hematological malignancies, which are not shared by their counterpart p300+/− mice.104,105 Taken together, this suggests that CBP and p300 are not redundant and may have some unique functions. 106
p300 and CBP function primarily as transcriptional cofactors for a number of nuclear proteins. They are able to interact with the basal transcription factors TATA-binding protein (TBP) and TFIIB,107,108 and can form a complex with RNA polymerase II.109,110 They are also cofactors for oncoproteins (e.g., fos, jun, and myb), transforming viral proteins (e.g., E1A) and tumor suppressor proteins (e.g., p53, E2F, Rb, or BRCA1). 111 Likewise, p300 and CBP aid transcription in 2 ways: firstly, they act as a bridge, linking DNA-binding transcription factors to the basal transcriptional machinery, and secondly, by acetylating histones, they create a permissive chromatin environment for gene expression.
Apart from transcription, p300 and CBP are also implicated in other processes. They interact with (and often acetylate) multiple proteins involved in DNA replication and repair, such as proliferating cell nuclear antigen (PCNA), DNA polymerase β, or thymine DNA glycosylase.45,112,113 CBP and p300 have been also implicated in the regulation of the progression of the cell cycle via interactions with cyclin E and cyclin-dependent kinase 2114-116 as well as in nuclear import by acetylation of importin-α. 117
Concomitant to their important physiological functions, p300 and CBP misregulation is strongly linked to cancer. p300 and CBP seem to have a dual role in oncogenesis; they can be either friends or foes. On the one hand, genetic studies show that they can act as tumor suppressors, as CBP- and p300-null chimeric mice develop hematological malignancies. 118 In fact, mutations of both HATs have been reported in human tumors; in particular, loss-of-function point mutations have been detected in colorectal, gastric, breast, ovarian, lung, and pancreatic carcinomas.119-121 On the other hand, aberrant activation or localization of those HATs can be oncogenic, and chromosomal translocations resulting in fusion proteins, including MLL-CBP, MLL-p300, MOZ-CBP, and MOZ-p300, have been identified in acute myeloid leukemia (AML), myeloid/ lymphoid or mixed lineage leukemia (MLL). 111 The oncogenic potential of MLL-CBP has been confirmed in murine models too. A tumor suppressor function for p300 and CBP is also contradicted by a number of cell culture studies, as depletion or reduction of those enzymes leads to proliferation defects and/or activation of senescence checkpoints. 111
Nonhistone Targets of Acetyltransferase Activities
Even though core histones are the best-characterized acetylation substrates, they are not the only ones. For example, high mobility group (HMG) chromatin-associated proteins have been shown to be acetylated already in the 1970s 122 ; however, the in vivo importance of these acetylation events still remains to be studied. On the other hand, posttranslational modification with acetyl groups has been shown to be important for the regulation of numerous transcriptional activators including the tumor suppressor p53, 123 proto-oncogene c-Myb, 124 tissue-specific activators such as GATA-1 or MyoD, 125 the cell cycle–related factor E2F, 126 or Tat, a transcriptional activator encoded by HIV-1. 127 Interestingly, many chromatin-modifying enzymes seem to be regulated via acetylation as well: not only are HATs and HDACs acetylated themselves,128,129 but also, the activity of the HMT SUV39H1 is regulated by acetylation in its catalytic SET domain 130 (see below). Protein acetylation takes place also outside the nucleus, in the cytoplasm and mitochondria, and it has been found to be a major mechanism by which key proteins are regulated in physiological processes such as cell migration, metabolism, and aging but also in pathological processes such as cancer and neurodegenerative disorders. 131 It is currently not known whether the cytoplasmic substrates of HAT/HDAC proteins are also involved in oncogenesis. A recent mass spectrometry screen identified 3,600 lysine acetylation sites on 1,750 proteins, 132 supporting the notion that acetylation is a widespread mechanism of regulating protein functions. 133 This also means that inhibiting HDACs does not only alter core histones’ acetylation but also acetylation of many other factors, which should be taken into consideration. Therefore, further studies are needed to uncover the truly important mediators of HDAC’s therapeutic effects.
Histone Phosphorylation
Histone phosphorylation is a dynamic modification in which a phosphate group is added to a specific amino acid’s residue. It was first identified in the late 1960s,134,135 and the first histone kinase was discovered in 1968. 136 For a long time, serine and threonine were the only residues identified to be phosphorylated in histones, but recent data also uncovered histone tyrosine phosphorylation (Fig. 2).137-141 Histones H1, H2A, H2B, H3, and H4 are all phosphorylated at multiple sites,142-147 but the most studied are so far the phosphorylations of H3. Histone phosphorylation reaction is catalyzed by several distinct kinases that are mostly specific for individual histone residues, although one histone residue can be phosphorylated by multiple kinases. 148
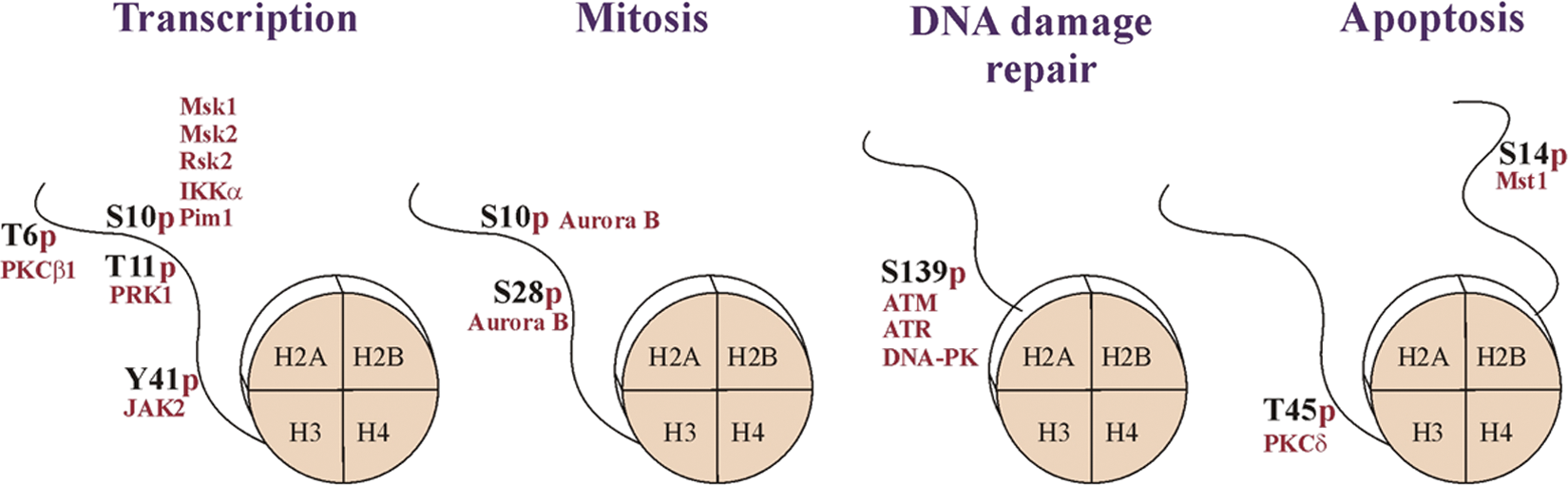
Histone phosphorylation is implicated in multiple cellular processes. Phosphorylation of the histone H3, H2AX, and H2B is involved in the regulation of transcription, mitotic chromatin condensation, and DNA damage response and apoptosis, respectively. Histone octamers and the modified tails are schematically shown. The enzymes responsible for these modifications (red) and the sites of phosphorylation are indicated. For details, see text.
The precise mechanistic role of histone phosphorylation in the regulation of chromatin structure is not yet fully elucidated. There are several potential mechanisms how histone phosphorylation can affect chromatin-related processes. For example, it has been shown that histone phosphorylation can directly influence chromatin compaction in vivo. 149 Studies in Tetrahymena demonstrated that formation of a “negative patch,” resulting from phosphorylation of many residues in linker histone H1, influences its binding to DNA and leads to an alteration in chromatin structure that facilitates transcription in this region. 149 Moreover, histone phosphorylation can also affect the binding of nonhistone proteins, excluding them from chromatin. The best-characterized example of this mechanism is the dissociation of HP1 protein from chromatin subsequent to mitosis-specific H3S10 phosphorylation (H3S10p). 150 In addition to this, phosphorylated histone residues can also be recognized by adapter proteins that serve as docking molecules for other nonhistone proteins.
Histone phosphorylation has been linked to many different cellular processes, for example, transcriptional activation, mitosis, meiosis, DNA damage repair, and apoptosis (Fig. 2). Therefore, a single phosphorylated residue within a histone can be linked to different chromatin states. For example, on one hand, H3S10p is associated with transcriptional activation of induced immediate-early genes, 151 but on the other hand, it is also globally present at the highly condensed chromatin during mitosis. 152
Histone Phosphorylation in Transcription Regulation
A number of studies demonstrate an essential role of histone H3S10 phosphorylation in gene activation. H3S10 is phosphorylated by various kinases. Phosphorylation of H3S10 by mitogen- and stress-activated protein kinases 1 and 2 (MSK1 and MSK2) as well as RSK2 kinase has been shown to play a role in the activation of mitogen-stimulated immediate-early response genes, such as c-fos and c-jun.146,151,153-155 H3S10 phosphorylation mediated by IκB kinase-α (IKK-α) occurs at NF-κB–responsive promoters in response to inflammatory cytokines. 156 Furthermore, Pim1 kinase catalyzes H3S10 phosphorylation at the E-boxes in Myc-target genes, contributing to their transcriptional activation after stimulation with growth factor. 157 It is noteworthy that PIM1 kinase was demonstrated to cooperate with Myc in cell transformation and tumor growth. 157
Additionally, an important role in transcriptional regulation can be found in a cross-talk between H3S10p and other histone modifications. For example, H3S10 phosphorylation can stimulate acetylation of histone H3K14,158,159 inhibit acetylation of histone H3K9, 160 and affect methylation of histone H3K9, 161 thus supporting an active transcriptional state. Gene activation by cooperative H3S10p and H3K14 acetylation has been broadly reported.158,162 In line with this simultaneous phosphorylation and acetylation, H3S10 and K14 can recruit 14-3-3 proteins concomitant with dissociation of heterochromatin proteins HP1 from adjacent H3K9me2. 163
Displacement of the HP1α protein from chromatin occurs not only due to H3S10p but also in response to H3Y41 phosphorylation. 141 This histone residue has been shown to be phosphorylated in hematopoietic cells 141 and is catalyzed by Jak2 kinase (Janus kinase 2), an enzyme that plays an important role in hematopoiesis and hematological malignancies.141,164 H3Y41 phosphorylation and displacement of HP1α lead to activation of oncogenes. 141 The genome-wide mapping of H3Y41-phosphorylated chromatin identified >2,000 potential JAK2-target genes, including genes encoding JAK2 itself, c-Myc, and the histone demethylase JMJD2C. 165
Another remarkable example of interdependence between histone H3 methylation and phosphorylation, which occurs between 2 nonadjacent sites within H3, comes from recent studies showing androgen receptor (AR)–dependent transcriptional regulation by protein kinase C–related kinase 1 (PRK1) and protein kinase C b I (PKCbI).166,167 Phosphorylation of H3T11 by the PRK1, in an androgen receptor–dependent manner, activates transcription by enhancing demethylation of H3K9 by the demethylases LSD1 (demethylating H3K9me1/me2) and JMJD2C (demethylating H3K9me2/me3). 166 PRK1 is crucial to androgen receptor function because PRK1 knockdown inhibits the androgen receptor–dependent transcription. It is noteworthy that high PRK1 and H3T11p levels correlate with high Gleason scores of prostate carcinomas, suggesting a role for PRK1 in tumorigenesis.
The PKCβ1-mediated phosphorylation of histone H3T6 plays also an important role in AR-dependent gene activation. In the presence of ligand, association of ligand-activated AR with PRK1 results in activation of PKCβI kinase that phosphorylates H3T6. Phosphorylation of H3T6 prevents the removal of H3K4 methyl marks by LSD1 (specific for H3K4me1/me2) and JARID1B (specific for H3K4me2/me3), which contributes to transcriptional activation. 167 Similar to H3T11p, the level of phosphorylated H3T6 also positively correlates with high Gleason scores of prostate carcinomas. 167 Moreover, knockdown of PKCβ1 can inhibit tumor cell proliferation in vitro and cancer progression of tumor xenografts in vivo, emphasizing the important role of PKCβ1 in the control of prostate tumor cell growth. 167
Histone Phosphorylation and Mitosis
Apart from transcriptional regulation, histone phosphorylation also plays essential roles during mitosis. Several histone sites have been detected to be phosphorylated by mitotic kinases and to be involved in chromatin condensation as well as in controlling chromosome localization of the proteins that function in chromosome segregation during cell division. A hallmark of mitosis is the global phosphorylation of H3S10 that begins during prophase, reaches maximum levels in metaphase, and is followed by a decrease during cell cycle progression to telophase. Reversible phosphorylation of H1 histones is also linked to mitosis. Phosphorylation of H1 is strongly cell cycle regulated, reaching maximum during late G2 and mitosis, and abruptly decreases in telophase. It occurs predominantly in the C-terminal tail on (S/T) PXK consensus motifs recognized by cyclin-dependent kinases, CDKs.168-171 Phosphorylation of both H1 and H3S10 specific for mitosis has been proposed to be required for proper condensation of mitotic chromatin.152,172 Mitotic H3S10 phosphorylation is mainly catalyzed by Aurora B. Apart from histones, Aurora B also phosphorylates a number of mitotic proteins. It is a catalytic component of the chromosomal passenger complex that regulates the spindle checkpoint and controls correct microtubule attachments to kinetochores.173-175 Although Aurora B has been found to be overexpressed in certain tumor types, its role in tumorigenesis is currently not clear. It is still not known whether the increased Aurora B expression in cancer cells reflects hyperproliferation or carcinogenesis. 176 Additionally, the evidence of genetic changes in AURKB locus in cancers is minimal. 177 On the other hand, increased Aurora B levels in several cancers including advanced colorectal cancer, glioblastoma, and ovarian and hepatocellular cancers have been associated with poor prognosis.178-180 Furthermore, exogenous overexpression of Aurora B in Chinese hamster embryo cells, concomitant with increased mitosis-specific H3S10 phosphorylation, can enhance chromosome instability and tumor invasiveness. 178
Histone Phosphorylation in DNA Repair
Histone PTMs, such as phosphorylation, ubiquitylation, and acetylation, play an essential role in DNA damage response pathways and participate in recruitment of downstream DNA damage response and repair proteins, as well as in the amplification of DNA damage signals. The histone H2A variant, H2AX, is a key component of DNA damage response.181,182 H2AX accounts for 2% to 25% of the total histone H2A pool in mammalian cells. 183 In response to DNA damage, H2AX is rapidly phosphorylated (i.e., within minutes) at S139, and this modification is spread over megabases from the break site. 184 Depending on the way double-strand breaks (DSBs) are induced, this reaction is mediated by several enzymes belonging to a family of phosphatidylinositol-3 kinase–like kinases (PIKK). Phosphorylation of H2AXS139 is catalyzed by ATM and DNA PK kinases in response to ionizing irradiation (IR)–induced breaks, and ATR governs H2AXS139 phosphorylation after DNA replication stress.185,186 The S139-phosphorylated H2AX, named γH2AX, plays an important role during the early stage of DNA damage response. It is necessary for the amplification of DNA damage signal and subsequent recruitment of a number of proteins involved in DNA damage response at the site of DSB. γH2AX is directly bound by MDC1 (mediator of DNA checkpoint 1), which serves as a platform for recruiting other DNA damage response proteins, including 53BP1 (p53-binding protein 1), BRCA1, NBS1 (Nijmegen breakage syndrome), and microcephalin.187-192 Subsequently, DNA damage response proteins transduce signals to Chk1 (checkpoint kinase 1) and Chk2 (checkpoint kinase 2) effector kinases that regulate many downstream targets, leading to the activation of DNA repair genes and a cell cycle delay. 193
Recent studies have shown that H2AX can also be phosphorylated at Y142.137,138 A novel kinase, WSTF (William syndrome transcription factor), is catalyzing this modification. H2AXY142 is constitutively phosphorylated at physiological conditions and is dephosphorylated by EYA (eyes absent homolog) phosphatases following the DNA damage.137,138 Dephosphorylation of H2AXY142p has been suggested to function as a switch mechanism determining cell fate after DNA damage, which enables the cell to choose between a DNA repair and an apoptotic response. 138 The proposed model is that H2AXY142 dephosphorylation, which occurs along with H2AXS139 phosphorylation (in response to ionizing irradiation), stimulates recruitment of proteins involved in DNA repair, such as MDC1, Mre11, and Rad50. 138 Conversely, when both H2AXS139p and H2AXY142p marks persist, apoptosis-inducing protein kinase JNK1 (Jun N-terminal kinase) is preferentially recruited and can stimulate an apoptotic response. 138 However, there are still unanswered questions concerning this hypothesis. For instance, whereas phosphorylation of S139 occurs very rapidly following DSB and reaches its maximum within minutes, the kinetics of H2AXY142p dephosphorylation are much slower and occur over several hours. 194 Further studies are needed to fully elucidate the role of Y142 phosphorylation and to completely understand the regulatory mechanisms to determine cell fate after DNA damage.
Considering the important role of H2AX phosphorylation in DNA damage response, it is not surprising that its deregulation is implicated in disease. Deficiency of both H2AX and DNA damage response proteins predisposes to cancer. The human H2AX gene (H2AFX) is located on chromosome 11q23.2-23.3, a region that is frequently lost in human cancers.195,196 It has been demonstrated that cells deficient for H2AX are sensitive to ionizing radiation and exhibit genomic instability and enhanced susceptibility to cancer.195,196 Furthermore, defects in kinases responsible for H2AX phosphorylation have been implicated in certain cancers. 197
Histone Phosphorylation in Apoptosis
Besides H2AXY142 phosphorylation, which has been suggested to function as a switch for apoptosis, the phosphorylation of histone H2B at S14 is also implicated in apoptosis. This modification is mediated by caspase-cleaved MST1 (mammalian STE20-like kinase 1) kinase. 198 During apoptosis, upon caspase-mediated cleavage, MST1 becomes cleaved, and its activated form translocates to the nucleus.199,200 The MST1-mediated H2BS14 phosphorylation (H2BS14p) correlates with chromatin condensation and occurs immediately prior to DNA laddering. Therefore, H2BS14p might be involved in the regulation of apoptotic chromatin condensation. Its mechanistic role is still unknown, but the authors propose H2BS14p to function through regulation of higher-order chromatin structure. 198 In support of this, phosphorylation of H2BS14 inhibits nuclear transport by regulating the regulator of chromosome condensation 1 (RCC1). Likewise, apoptosis-related phosphorylation has also been detected to occur on histone H3T45, catalyzed by protein kinase Cδ (PKCδ). 201 The functional role of H3T45 phosphorylation has not yet been elucidated; however, it has been suggested that it might induce structural changes within the nucleosome, thus enabling DNA fragmentation.
Histone Methylation
Methylation of histone tails occurs mainly at lysines (K) and arginines (R). In this review, we focus mainly on lysine methylation; for a recent review on arginine methylation, see Di Lorenzo and Bedford. 202 Although, in the early days, histone methylation was considered to be a stable and irreversible epigenetic mark, the discovery of the lysine histone demethylases (HDMs) flipped over this idea and showed that it can be dynamic. 203 This finding also provided the first indication of how methylation may regulate transcription and enable gene maintenance in an on or off state.
Methylation as a histone PTM has a number of fundamental differences compared to the above-described acetylation and phosphorylation. Lysines can be monomethylated, dimethylated, or trimethylated, whereas arginines can be monomethylated or dimethylated. For arginine methylation, symmetric or asymmetric dimethylation must be distinguished because the precise methyl states are relevant for functional outcome. In contrast to acetylation, addition of a methyl group on a side chain does not alter its charge, and therefore, it is unlikely that methylation would directly modulate nucleosomal interactions required for chromatin folding. Another distinct characteristic of histone methylation is that it has been implicated in both transcriptional activation and repression. For example, trimethylation of H4K20 (H4K20me3), H3K9, and H3K27 was shown to be involved in transcriptional silencing, whereas trimethylation at H3K4 (H3K4me3) and H3K36 act as active marks. 15 Arginine methylation, catalyzed by a family of protein arginine methyltransferases (PRMTs), can act as a transcriptional activator or repressor. 204 Methylation of lysines has been described on all 4 core histones but also on linker histone H1. H1.4K26 was the first methylation site studied in H1. 219 Recently, H1.2K187 has been identified as a novel G9a target on H1, 205 and multiple other methylation sites have been mapped on the linker histone, awaiting functional characterization. 11
It is now well known that lysine methylation can provide a binding surface that coordinates the recruitment of histone “readers.” Hence, multiple protein domains binding specifically to methyl lysine had been identified: PHD fingers (for binding to H3K4me3 as in ING2 and the BPTF PHD finger of NURF),206,207 Tudor domains (for binding to H3K4me3 and H4K20me3 as in JMJD2A demethylase), 208 chromodomains (for binding to H3K9me2/3 as in HP1), 209 and WD-40 domains (for binding to H3K27me3 as in EED).210,211 Proteins containing these domains can be found in many chromatin-remodeling and chromatin-modifying complexes as well as transcriptional activators/repressors. These specific “code-reading” abilities can help to either maintain or also rapidly rearrange local genomic regions in an active or inactive chromatin state. lysine methylation may also regulate the depletion of specific methyl-sensitive binders that interact with unmethylated histone tails 212 and by that may also prevent the propagation of adjacent modification on a following residue.
Taken together, because methylation has so many faces and is considered to be an essential step involved in many cell fate determinations such as developmental and differentiation processes,213-215 pluripotency, 216 and maintaining genome integrity, 217 it is no surprise that it is highly associated with cancer and other diseases (approximately 50% of the HMTs encoded by the human genome are now linked to diseases and in particular tumorigenesis). In the following part, we mainly focus on HMTs that have clear links to tumorigenesis and discuss the possible mechanisms and pathways by which histone methylation may contribute to cancer formation.
The Histone Methyltransferase EZH2, a Key Component of the Polycomb System
Enhancer of zeste homolog 2 (EZH2) is one of the best-studied HMT enzymes involved in oncogenesis. It is an evolutionarily conserved transcriptional repressor that specifically dimethylates and trimethylates H3K27 residues. 218 In addition, along with another HMT G9a, it has also methyltransferase activity towards the linker histone H1.4K26.219,220 EZH2 belongs to the Polycomb repressive complex 2 (PRC2), which is 1 of the 2 classes of Polycomb group proteins (PcG), originally discovered in fruit flies and involved in the silencing of the HOX gene cluster. 218 Although EZH2 is the active enzyme within PRC2, it requires the other members of the complex, suppressor of zeste 12 (SUZ12) and embryonic ectoderm development (EED), for its enzymatic activity. 214 Through targeted trimethylation of H3K27 at regulatrory regions in the genome (and potentially other targets), Polycomb group proteins affect a wide range of cellular processes that includes apoptosis, proliferation, differentiation, and cell cycle regulation.221,222 In mammals, Polycomb group gene expression is important in many aspects of development. It is required for early mouse development214,223 as well as for maintaining ES cell pluripotency and plasticity during embryonic development.224,225 Moreover, null mutations for the PRC2 genes SUZ12 and EZH2 lead to embryonic lethality in mice.214,223 Currently, there are numerous lines of evidence suggesting that Polycomb group proteins are dysregulated in cancer and may promote cancer progression. 222 Indeed, EZH2 is found to be overexpressed in many types of aggressive cancers such as the breast, lung, liver, nasopharyngeal, colon, and prostate as well as in many other types of carcinomas.226-229 Moreover, EZH2 can transform the growth of a normal prostate epithelial cell line both in vitro and in vivo. 230 These findings show that EZH2 can potentially act as an oncogene rather than simply be a marker for hyperproliferation. Furthermore, RNAi-targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer, blocks prostate cancer metastasis, significantly decreases breast xenograft growth in addition to improved survival in breast cancer models, and reduces proliferation of human papillomavirus-positive tumor cells.231-234
Despite the fact that the mechanism of EZH2 in oncogenesis is not yet clear, it is believed that H3K27me3 methylation of promoters of tumor suppressor genes may be involved in their anomalous silencing. Indeed, it has been shown that EZH2 represses the expression of several tumor suppressor genes, including p16 INK4a, 228 E-cadherin, 235 DAB2IP, 232 RUNX3, 236 BRCA1, 231 and the adrenergic receptor β2, which is expressed at low levels in metastatic prostate cancer and tumors that exhibit poor prognosis. 237 Additionally, oncoproteins can activate EZH2 to induce epigenetic silencing. 238 In contrast to EZH2 activation, several inactivating mutations of EZH2 have been recently discovered in myeloid malignancies, suggesting that EZH2 can also act as a tumor suppressor. 239 Likewise, recurrent EZH2 mutations were described in lymphomas of germinal center origin. These mutations replace a single tyrosine in the catalytic SET domain of the EZH2 protein (Y641) and reduce its enzymatic activity. 240 Together, these findings show that although inhibition of EZH2 was proposed as a therapeutic strategy, inactivating EZH2 might promote other malignancies. Despite what we described above, it is still unclear whether EZH2 overexpression is a cause or a consequence of tumorigenesis. Dysregulation of EZH2 in cancer may well occur as a result of a secondary effect due to the high proliferative nature of solid tumor and metastatic cells, for instance, through the regulation of microRNAs (miR), which are known to regulate multiple target genes. Indeed, miR-101 microRNA was found to target EZH2 but also to regulate cell proliferation, invasion, and tumor growth. Interestingly, a loss of this miRNA leads to overexpression of EZH2 in several cancers. 241 In addition, miR-26a, which is downregulated in breast cancer specimens and cell lines, was also found to target EZH2. Upon miR-26a downregulation, EZH2 becomes significantly upregulated in breast cancer. 242
In line with the concept of histone modifiers being themselves regulated by PTMs, CDK1/2-dependent phosphorylation at multiple residues (T345, T350, and T487) can regulate EZH2 activity and levels.25,26 While phosphorylation at T345 increases the binding of EZH2 to the HOTAIR noncoding RNA that may facilitate its recruitment to target genes, the phosphorylation at T487 disrupts EZH2 binding to other PRC2 components SUZ12 and EED and thereby inhibits its methyltransferase activity, resulting in inhibition of cancer cell invasivness.25,243 Moreover, phosphorylation of EZH2 at T350 is important for its recruitment and maintenance of H3K27me3 levels at EZH2 target loci. Therefore, blocking T350 phosphorylation not only diminishes the global effect of EZH2 on gene silencing but also mitigates EZH2-mediated cell proliferation and migration. 244
Altogether, this clearly links EZH2 with oncogenesis and demonstrates that accurate and precise regulation of H3K27 methylation is needed in order to prevent cancer development and progression.
Histone H3K9 Methyltransferases
In concordance with other methyl-lysine marks, H3K9 monomethylation, dimethylation, or trimethylation can induce distinct chromatin states. H3K9me3 was found to correlate with transcriptional silent chromatin and to act as a specific binding platform for the heterochromatin protein 1 (HP1). 17 The SUV39H1 and SUV39H2 enzymes specifically trimethylate H3K9 and use monomethylated H3K9 as a preferred substrate. By doing so, they play a critical role in the establishment of constitutive heterochromatin especially at pericentric heterochromatin. 245 The combined disruption of both Suv39h genes severely impairs mice viability and induces chromosomal instabilities (primarily by impairing chromosome segregation) together with an increased risk of developing B cell lymphomas. 246 It seems that H3K9 methylation protects cells from genome instability, which is a critical pathway in cancer progression, and its absence can result in tumorigenesis. Additionally, SUV39H1 is also involved in the repressive functions of the retinoblastoma (Rb) tumor suppressor protein, which targets the enzyme to promoters of cell cycle control genes. 247 Moreover, the HDAC SirT1 interacts with the N-terminal domain of SUV39H1 and deacetylates SUV39H1 on K266 to stimulate its methyltransferase activity. The binding of SirT1 alone can also activate SUV39H1 through additional mechanisms independent of its deacetylase activity. Therefore, the SUV39H1-SirT1 complex may regulate transcriptional silencing through the HDAC activity of SirT1 and the enhanced methylase activity of SUV39H1. 130 Interestingly, recent studies show that DBC1 (deleted in breast cancer 1) not only binds to and inhibits SirT1 but also binds to the catalytic domain of SUV39H1 and causes its complete inactivation. 248
The HMT G9a (and the HMT Glp1, an interaction partner of G9a) is classified as a H3K9 methyltransferase. Indeed, H3K9 methylation levels decrease in G9a-deficient embryos, which displayed severe growth retardation and early lethality. 249 However, recently, it had been shown that G9a could also contribute to H3K27 (H3K27me1 and H3K27me2) methylation in vivo. 250 G9a was found to promote lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. 251 Besides this, G9a was upregulated in aggressive lung cancer cells, and its elevated expression correlated with poor prognosis. Additionally, RNAi-mediated knockdown of G9a in highly invasive lung cancer cells inhibited cell migration and invasion in vitro and metastasis in vivo. 251
Histone H3K4 Methyltransferases
A well-studied example of a H3K4-specific HMT involved in human cancer is the mixed lineage leukemia (MLL) protein. MLL is a mammalian trithorax group (Trx-G) protein that regulates Hox gene expression and counteracts PcG function. Rearrangements of the MLL gene, which is located at chromosome 11q23, are associated with aggressive acute leukemias in both children and adults and include MLL fusion genes, partial tandem duplications, or amplification of MLL. 252 Whereas MLL-deficient mice are embryonic lethal and have defects in the hematopoietic system accompanied by Hox gene dysregulation,253,254 mice carrying MLL fusions and amplifications often upregulate Hox gene expression. They develop acute myeloid leukemia that apparently results in a block of hematopoietic differentiation. Therefore, MLL fusion proteins are thought to contribute to tumorigenesis by a gain-of-function mechanism rather than a loss-of-function mechanism. 255 MLL tumorigenic activity is not necessarily related to its HMT activity because the loss of the SET domain in MLL1 N-terminal fusions upregulates the expression of several target genes, which are essential for the transforming activity of the MLL fusion proteins. 256
The SET and MYND domain– containing protein 3 (SMYD3) specifically methylates H3K4 and plays an important role in transcriptional regulation as a member of the RNA polymerase complex. 257 Through the interaction with the RNA helicase HELZ, SMYD3 can form a complex with RNA polymerase II and activates a set of genes that includes oncogenes, homeobox genes, and genes associated with cell cycle regulation. Concordantly, it can be commonly found upregulated in colorectal and hepatocellular carcinomas. 257 Overexpression of SMYD3 enhances cell growth, whereas siRNA in cancer cells results in growth suppression. 257 A parallel study from the same group shows that SMYD3 expression is elevated in a great majority of breast cancer tissues and its increased levels are essential for proliferation of the breast cancer cells. 258 Additionally, SMYD3 can promote breast carcinogenesis by directly regulating the expression of the proto-oncogene WNT10B. In another study, it was found that human cancer cells express both the full-length and a cleaved (apparently proteolytic cleavage of the full-length protein) SMYD3 protein, resulting in a form of SMYD3 protein that lacks 34 amino acids in the N-terminal region and has a higher HMTase activity compared to the full-length protein. 259 This indicates that the N-terminal region of SMYD3 plays an important role in the regulation of its enzymatic activity and may serve as an attractive target to abolish SMYD3 methyltransferase activity.
Modifying the Modifiers by Methylation
Apart from histones, chromatin-modifying enzymes can also be methylated by HMTs and their activity modulated by this methylation. A recent example for this is the regulation of DNA methyltransferase-1 (DNMT1) stability through SET7-mediated lysine methylation (Fig. 3A). The HMT SET7 (a H3K4- specific HMT) directly interacts with DNMT1 and specifically monomethylates DNMT1 at K142. This methylation peaks during the S and G2 phases of the cell cycle and promotes DNMT1 degradation. 260 Moreover, as part of a complex cross-talk as shown for histones, the interplay between monomethylation of DNMT1 K142 by SET7 and phosphorylation of DNMT1 S143 by AKT1 kinase can regulate DNMT1 stability by a methylation/phosphorylation switch. 261
Similar to histones, p53 is subject to both activating and repressing lysine methylation (Fig. 3B). For instance, SMYD2 monomethylates p53 K370 (K370me1), which represses its activity. 262 On the other hand, p53 K370 dimethylation (K370me2) was shown to promote the association of the coactivator p53-binding protein 1 (53BP1) through the tandem Tudor domains in 53BP1. Thus, by removing this methylation, the lysine-specific demethylase LSD1 represses p53 function through the inhibition of interaction of p53 and 53BP1. 263 Likewise, the Set domain–containing protein Set9 monomethylates p53 at K372 (K372me1), resulting in the nuclear localization and increased stability of p53. 264 In addition, Set9-mediated methylation of K372 was shown to inhibit Smyd2-mediated methylation of K370. All together, this demonstrates that p53 is dynamically regulated by lysine methylation and demethylation and that the methylation status may produce different regulatory outputs. The example of p53 shows that a similar regulatory complexity can exist for nonhistone proteins as for histones.265,266
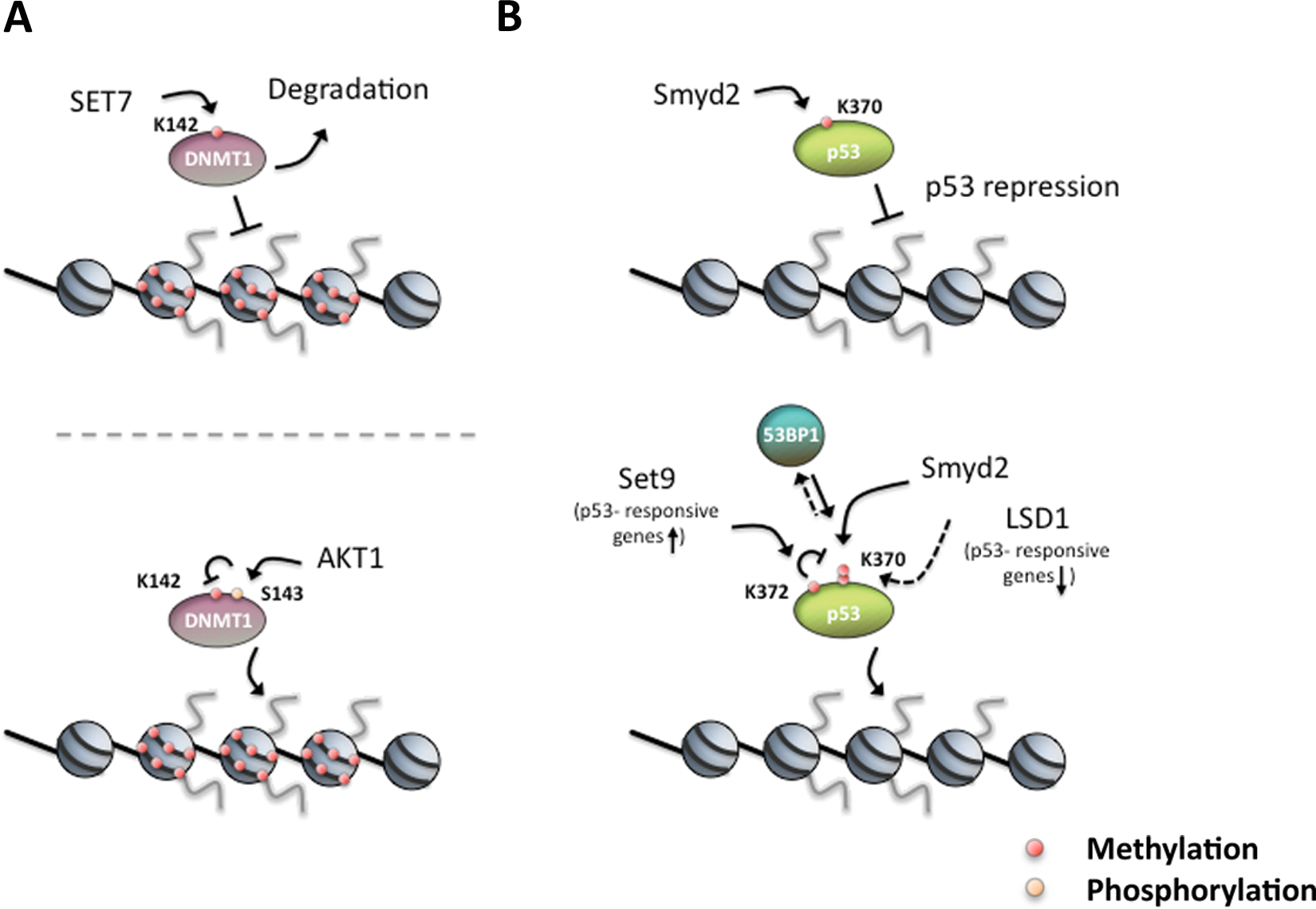
Nonhistone proteins are dynamically regulated by PTMs to control their activity. (
Concluding Remarks and Perspective
Although in the last decades the chromatin field provided a wealth of knowledge about the biology and function of histone modification together with its regulating mechanisms, the exact impact and influence of certain specific modifications on the tumorigenic process are still largely unclear. Several lines of evidence suggest that a dysregulation of the epigenetic machinery contributes to human cancers through disturbing the “histone code” and/or its readout, but it remains to be determined whether the dysregulation of either the epigenetic players or disruption of specific marks is the initiator or just the outcome effect of oncogenesis.
Histone modifications and their modifiers hold great promise as therapeutic targets because, in contrast to genetic mutations, PTMs are dynamic and potentially reversible. On top of that, many histone modifications are set by site-specific modifiers at a precise and targeted position, thereby most suitable for inhibition. Indeed, although we do not yet fully understand the underlying mechanism, HDAC inhibitors are proven to be highly effective anticancer drugs that inhibit tumor growth, reactivate tumor suppressor genes, and lead to genomic instability by a variety of mechanisms. In line with histone acetylation as a potential therapy target, a recent article showed that inactivating mutations of acetyltransferase genes act as a major pathogenetic mechanism shared by common forms of B cell non–Hodgkin lymphoma, suggesting that reduction in HAT dosage is important for lymphomagenesis. 267 Interestingly, this work also demonstrates specific defects in acetylation-mediated inactivation of the BCL6 oncoprotein and activation of the p53 tumor suppressor proteins. 267 In addition to HDAC inhibitors, methyltransferase inhibitors have now been broadly used for cancer, inflammatory, and autoimmune diseases. 268
We are currently only beginning to understand the diverse implications of histone PTMs and the corresponding modifying enzymes for biology and about disease. We do not know enough how PTMs integrate in normal biological processes and networks, for example cell proliferation, differentiation, or development, and therefore, we do not have enough insights into their deregulation in diseases such as cancer. The significance of studying posttranslational modifications extends beyond the field of chromatin research because changes in the modification patterns are likely to affect many aspects of protein functions. Therefore, apart from their role in chromatin function, it is important to see histone modifications as a model system to understand how covalent modifications regulate protein functions.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported by the Max Planck Society; the Deutsche Forschungsgemeinschaft [grant number SFB 746]; and a European Research Council starting grant. The Polish State Committee funds E.P. for Scientific Research [grant number NN303813140]. I.C. is a Humboldt fellow.