
Abstract
Normal cellular behavior can be described as a complex, regulated network of interaction between genes and proteins. Targeted cancer therapies aim to neutralize specific proteins that are necessary for the cancer cell to remain viable in vivo. Ideally, the proteins targeted should be such that their downregulation has a major impact on the survival/fitness of the tumor cells and, at the same time, has a smaller effect on normal cells. It is difficult to use standard analysis methods on gene or protein expression levels to identify these targets because the level thresholds for tumorigenic behavior are different for different genes/proteins. We have developed a novel methodology to identify therapeutic targets by using a new paradigm called “gene centrality.” The main idea is that, in addition to being overexpressed, good therapeutic targets should have a high degree of connectivity in the tumor network because one expects that suppression of its expression would affect many other genes. We propose a mathematical quantity called “centrality,” which measures the degree of connectivity of genes in a network in which each edge is weighted by the expression level of the target gene. Using our method, we found that several SRC proto-oncogenes LYN, YES1, HCK, FYN, and LCK have high centrality in identifiable subsets of basal-like and HER2+ breast cancers. To experimentally validate the clinical value of this finding, we evaluated the effect of YES1 knockdown in basal-like breast cancer cell lines that overexpress this gene. We found that YES1 downregulation has a significant effect on the survival of these cell lines. Our results identify YES1 as a target for therapeutics in a subset of basal-like breast cancers.
Introduction
Almost 200,000 cases of invasive breast cancer are diagnosed each year in the United States, and of these, over 40,000 women die each year from this disease. 1 It is well established that breast cancer is not a single disease but consists of several types with distinct clinical behaviors and treatment approaches. Approximately 60% to 70% of tumors express the estrogen receptor (ER) and are treated by drugs targeting the estrogen signaling pathway. 2,3 Approximately 20% to 30% have amplification of the human epidermal growth factor receptor-2 (HER2+) and are treated with Herceptin (Genentech Inc., South San Francisco, CA) and other agents that target the HER2 transmembrane receptor tyrosine kinase. 4 However, there remains significant heterogeneity in both natural history and treatment response in tumors with similar clinical classification. 2-4
Gene expression analyses have provided insight into this clinical heterogeneity. Supervised learning methods applied to gene expression data have identified gene panels predictive of risk that are currently being applied to clinical practice. 5,6 Unsupervised clustering of gene expression data 7,8 has identified further stratification of breast cancer into subtypes with distinct gene expression profiles correlated with recurrence risk and survival. These studies divide breast cancers into luminal A (ER+ with good prognosis), luminal B (ER+ with poor prognosis), HER2+ (HER2+, ER−), and basal-like (HER2−, ER−). These subtypes have been validated in several subsequent studies. 9,10
Basal-like breast cancers (BLC) are high-grade, invasive cancers that are characterized by the “triple-negative” phenotype (ER−/PR−/HER2−), lacking expression of the estrogen receptor (ER), progesterone receptor (PR), and HER2. BLC account for approximately 15% of human breast cancers, tend to occur in younger women, and account for a disproportionate amount of breast cancer deaths. 11 As BLC do not respond to either hormonal treatment or HER2- targeted treatment, there are at present no targeted therapies for this aggressive cancer. The goal of our work is to try to identify specific therapeutic targets for triple-negative breast tumors. Towards this end, we hypothesized that good targets should be highly expressed proteins that are “important” for the survival of the tumor cell, meaning that downregulation of the associated gene would lead to a significant impact on the fitness of the cancer cells. Intuitively, the expression levels of such important genes should be correlated with a large number of other genes across subsets of samples. In effect, these genes are hubs (high-degree nodes) in the associated gene network. Simulations performed on synthetic gene networks 12 have shown that knocking out a highly connected gene (a gene linked to many other genes) has a greater impact on fitness compared to knocking out a gene with fewer connections, and this effect is even stronger for genes with high expression values.
Using these ideas, we have developed a new method for identifying therapeutic targets in different cancer types from gene expression data. In our method, the usual measurements of differential expression are replaced by a new paradigm called “gene centrality.” The identification of a target gene as one with high centrality is based on our expectation that in addition to being overexpressed, good therapeutic targets must have a high connectivity degree in the tumor gene network. We identify such genes by computing the eigenvector associated with the largest eigenvalue of a modified gene connectivity network matrix in which each edge is weighted by the overexpression level of the target gene. Genes with high centrality are identified as those with high coefficients in the eigenvectors with the highest eigenvalues, which represent the majority of variation in the data. We expect such genes to be better therapeutic targets because their modification affects a number of other “important” genes.
We applied this method to published breast cancer gene expression datasets. Classification of tumors into luminal, HER2+, and basal-like breast cancer subtypes was performed using consensus clustering and centroid-based classification as described in Alexe et al. 13 Genes with high centrality scores for each subtype were identified using the network analysis as described above. This led to the identification of a number of SRC tyrosine kinases (LYN, YES1, HCK, FYN, and LCK) with high centrality scores in subsets of basal-like breast cancers and in HER2+ tumors. In this article, we focus our analysis on the YES1 kinase, which we found to be associated with the basal-like subtypes. The importance of YES1 as a potential therapeutic target was verified via a growth/survival assay by stably suppressing the expression of YES1 in several breast cancer cell lines. We showed that downregulation of YES1 had a significant effect on the fitness of the cancer cells. This analysis suggests that several current drugs, including SRC inhibitors, may be successfully used to treat a molecularly identifiable subset of basal-like breast cancer patients.
Results
Gene expression datasets
We analyzed gene expression data from Wang et al., 14 consisting of 286 early-stage, lymph node–negative breast tumor samples from patients treated with surgery and radiation but no adjuvant or neoadjuvant systemic therapy. Robust, unsupervised consensus clustering previously applied to this dataset has identified 6 core breast cancer subtypes, 13,15,16 2 within each clinical subtype: luminal (ER+) cases split into 28 luminal A (LA) and 104 luminal B (LB) samples, HER2+ (HER2+/ER−) cases split into 14 HER2I and 17 HER2NI, while basal-like (ER−/HER2) cases split into 15 BA1 and 22 BA2 samples. LA and LB tumors are both ER+ and PR+, with the main difference between them being that LB cases had a significantly higher recurrence rate. Compared to BA2, the basal-like subtype BA1 was characterized by overexpression of genes associated with the innate immune/defense response pathway (for details, see Alexe et al. 13 ). The relationship of BA1 and BA2 subclasses of BLC to the “claudin-low” subset of triple-negative breast cancers recently described using the “intrinsic gene set” is not clear. 40 HER2I and HER2NI breast cancers both have amplification of the ERBB2 (HER2) gene. However, the HER2I subtype is distinguished from HER2NI by a strong upregulation of lymphocyte-associated genes and an infiltration of lymphocytes into the tumor, which is easily seen on pathological examination of FFPE slides. 13 Moreover, HER2I patients have a significantly lower recurrence rate compared to HER2NI tumors in the absence of adjuvant therapy. 13
In addition, we also analyzed another gene expression dataset from Ivshina et al., 17 consisting of 249 samples from primary invasive breast tumors. Samples were classified into subtypes using the core clusters already identified in the Wang et al. 14 dataset, by comparing gene expression values for each sample to mean expression values calculated for each of the original core clusters. Centroids for each subtype were identified using normalized gene expression values as described in Alexe et al., 13 and distances from the centroids to samples from the new dataset were calculated using several metrics (such as Pearson correlation and Euclidean distance). For each distance metric used, the new samples were assigned to the subtype whose centroid they were closest to. Samples that did not consistently classify with the same subtype for all distance measures were discarded. We thus identified 78 LA, 96 LB, 12 HER2I, 24 HER2NI, 11 BA1, and 13 BA2 tumors in the Ivshina et al. dataset. 17
Both gene expression datasets were downloaded from the Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) database. Accession identifiers GSE2034 and GSE4992 correspond to the first and second dataset, respectively. Table 1 summarizes the clinical and pathological characteristics of all patients used in the study. The main difference between the 2 datasets is in the distribution of lymph node (LN) status and histological grade. However, this does not adversely affect our analysis because it depends mostly on the subtype assignment, which is unambiguous (Table 1).
Microarray Datasets Used in This Study
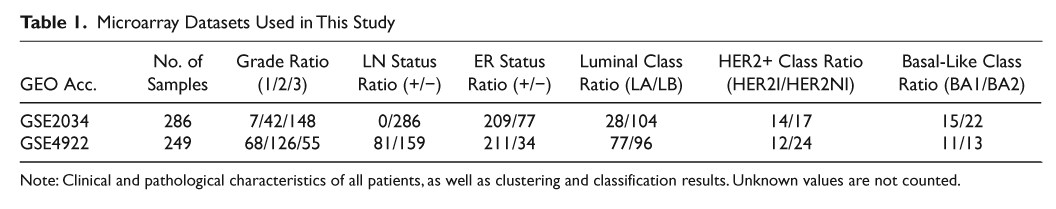
Note: Clinical and pathological characteristics of all patients, as well as clustering and classification results. Unknown values are not counted.
High centrality score genes in robust breast cancer subclasses
The gene expression data were used to compute outlier scores (θ) and Pearson correlation values (r) for each gene across all samples. Centrality scores were then computed as described in the Materials and Methods section to identify oncogenes with high centrality scores in each breast cancer subtype. As seen in Table 2, our method successfully identified ERBB2 in HER2+ (HER2I, HER2NI) subtypes and ESR1 for luminal subtypes (LA, LB) as high centrality genes that are potential therapeutic targets. Most strikingly, our methods found a set of related SRC protein kinases LYN, YES1, HCK, FYN, and LCK with high centrality scores in the BA1 subset of basal-like breast cancer. In addition, all these kinases except YES1 also have high centrality scores in the HER2I subset of HER2+ breast cancers. Our analysis therefore suggests that these specific kinases are all potential therapeutic targets for patients in these specific subtypes of breast cancer.
Top Centrality Results for Cancer Genes
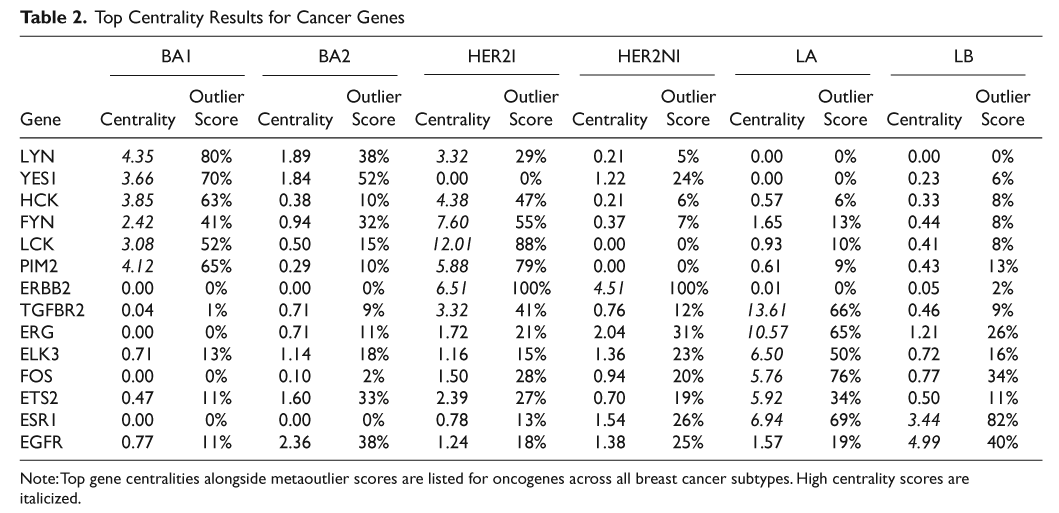
Note: Top gene centralities alongside metaoutlier scores are listed for oncogenes across all breast cancer subtypes. High centrality scores are italicized.
In luminal A cancers (mostly low-grade ER+ breast cancers), in addition to the estrogen receptor-alpha (ESR1), genes with high centrality scores included ERG, ETS2, and ELK3 that belong to the ETS family of transcription factors, TGFBR2, and the tyrosine kinase FOS. In luminal B cancers, EGFR had high centrality scores.
YES1 in basal-like breast cancers
To test whether centrality scores can successfully identify novel therapeutic targets in basal-like, we focused on the YES1 (Yamaguchi sarcoma viral oncogene homolog 1) gene. This gene is known to be also overexpressed in colorectal, head and neck, renal, lung, and stomach cancers. 18 Figure 1A and 1B show the normalized expression values of YES1 across all subtypes for the 2 datasets. To avoid sampling bias (due to an unequal number of samples in the subtypes), we used the following procedure: 10 samples were chosen from each subtype, the expression value of YES1 was standard normalized across these 60 samples, and the normalized value of each sample in this bootstrap dataset was noted. This procedure was repeated 1,000 times. The average expression value of YES1 for each sample across these bootstrapped datasets was calculated, keeping track of how often the sample appeared in the bootstrap samplings. Figure 1A and 1B shows the sorted values of YES1 for all samples in each subtype thus obtained for the 2 datasets. The relative overexpression of YES1 in the basal-like subtypes is obvious from Figure 1A and 1B. We note that YES1 is also relatively overexpressed in HER2-NI. The centrality score of YES1 in HER2-NI was >1 and was higher than all other related SRC kinases but still fell below the threshold of centrality set for the whole dataset.
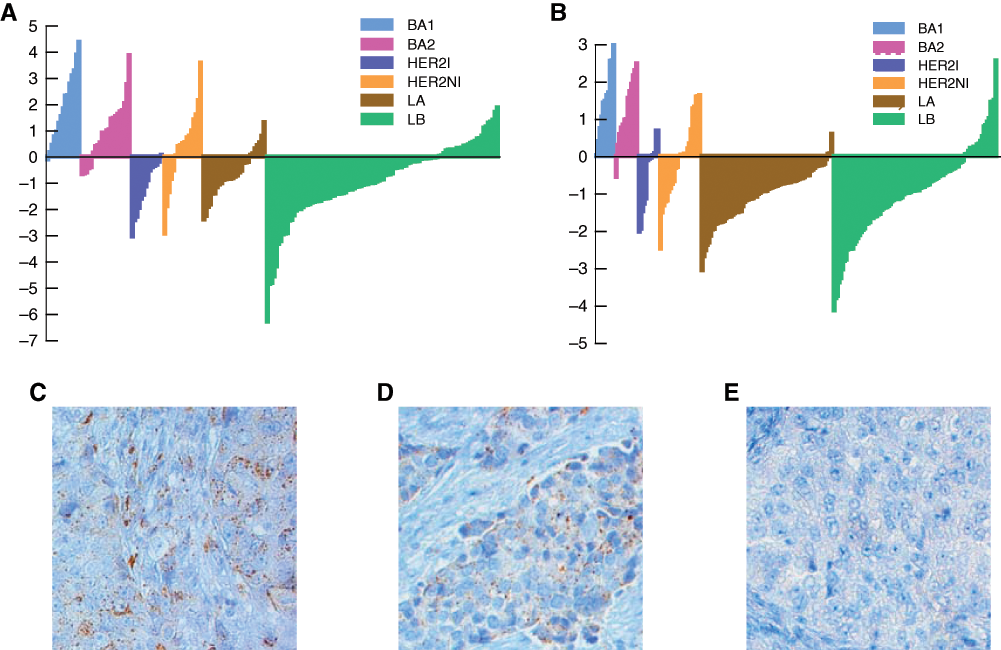
YES1 is overexpressed in a subset of basal-like breast tumors. Bar plots showing relative overexpression of YES1 in a subset of basal-like breast tumors in the GSE2034 (
To further validate YES1 overexpression in a subset of basal-like breast cancers, we analyzed FFPE slides from 13 ER−/PR−/HER2−, high-grade breast cancers with an invasive ductal morphology. No gene expression data were available for these clinical samples. Although BLC can only be formally identified by gene expression profiling, published data have demonstrated that >85% of high-grade ER−/PR−/HER2− breast cancers with invasive ductal morphology will fall into the BLC category by gene expression profiling. 41 Thus, we expect this cohort of 13 triple-negative breast cancers to consist mostly of BLC. Tissue slides were obtained under an IRB-approved protocol from the Tumor Bank at the Cancer Institute of New Jersey. Immunohistochemical analysis of the slides was performed using a YES1-specific antibody and scored as described in the Materials and Methods section.Figure 1C, 1D, and 1E show staining of the samples identified as having high, medium, or low/no expression of YES1, respectively. Of the 13 samples, 2 showed high levels of YES1, 6 had medium expression, and 5 had low/no expression. These data demonstrate that a subset of triple-negative breast cancers has high expression of YES1 and support the hypothesis that YES1 is overexpressed in a subset of BLC.
Validating YES1 as a therapeutic target in a subset of basal-like breast cancers
The analysis described above supports the hypothesis that YES1 is overexpressed in a subset of basal-like breast cancers and hence is a possible therapeutic target. We tested this hypothesis by determining whether suppressing YES1 expression in subsets of breast cancer cell lines has a significant effect on cell survival/growth. Lentiviral vectors expressing YES1-specific shRNA were constructed to stably suppress the expression of YES1 in breast cancer cell lines: MDA468, MDA231, BT549, MCF10A, SKBR3, and MCF7. Of these, MDA468, MDA231, BT549, and MCF10A are all basal-like; that is, they are negative for expression of estrogen, progesterone, and HER2 proteins (ER−/PR−/HER2−) and have been shown to profile as “basal-like” by gene expression analysis. 42 SKBR3 is ER−/PR− but HER2+, while MCF7 is ER+/PR+ and consistent with the luminal breast cancer type. Three different shRNA were chosen, and their ability to suppress the expression of YES1 was tested on MDA468. Only the most efficient one (shYES1#2) was selected for subsequent experiments. Equal numbers of cells from each cell line were infected with either a lentivirus-encoding shYES1 or a control-scrambled shRNA. After 6 days, all cells were counted and the results compared to the controls to assess whether the growth rate of cancer cells was affected by silencing YES1. We found (Fig. 2A, 2B, and 2D) that all cell lines except the luminal cell line MCF7 had a significant reduction of cell counts when treated with shYES1#2 compared to the controls. Two additional shRNAs that were less efficient in knocking down YES1 protein levels (shYES1#1 and shYES1#3) could also decrease cell growth in this assay, although not as efficiently as shYES1#2, demonstrating the effect on growth is not likely an off-target effect of shYES1#2 (Fig. 2C).
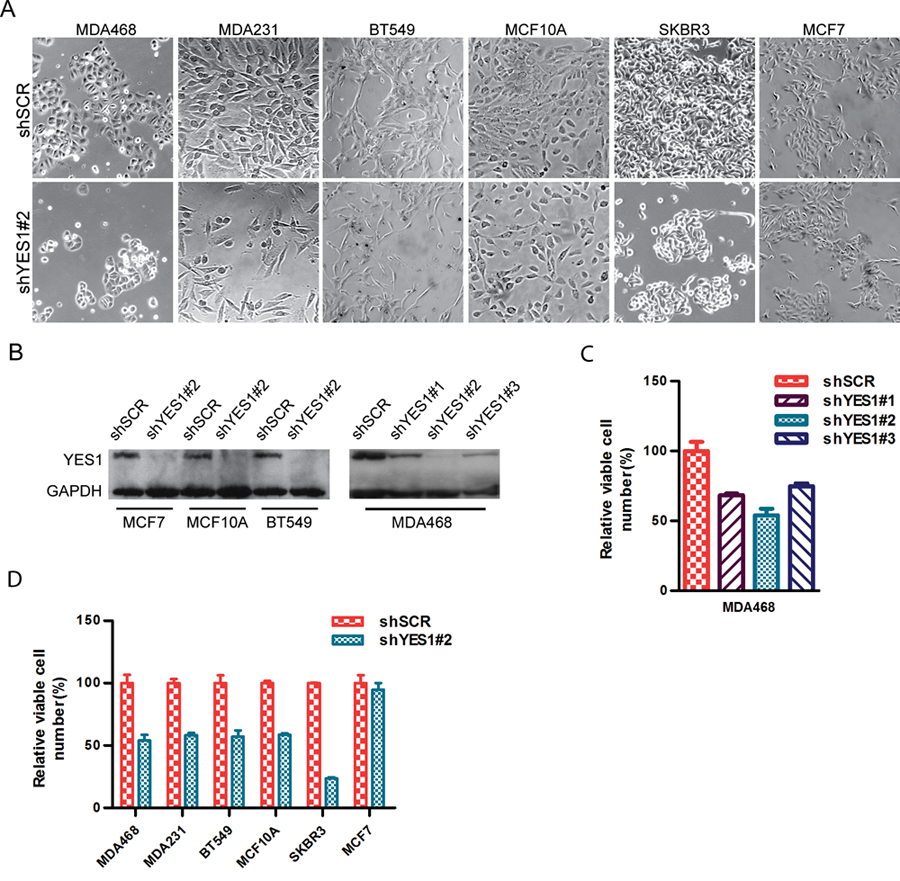
YES1 knockdown impairs the growth of breast cancer cell lines. A series of 6 breast cancer cell lines were infected with lentiviral constructs designed to suppress expression of YES1 with hairpin shRNA. Equal numbers of these cells were plated in triplicates alongside controls and then counted after 6 days. (
Discussion
In this article, we have developed and applied a novel method for analyzing gene expression data that takes into account not only the levels of expression for different genes but also a measure of similarity between pairs of genes across subsets of samples in subtypes. We have introduced a novel analytical measure called “gene centrality,” which identifies potential therapeutic targets in subsets of cancer patients. The algorithm implemented here, based on outlier scores and correlations, is general and can be modified for the analysis of any cancer. It can also be extended using different measures of expression, as long as they are positive, and other normalization schemes (such as the soft-max normalization procedure described in Han and Kamber 19 ). Other estimations of correlation, such as the Spearman rank correlation, 20 Kendall tau rank correlation, 21 or Mutual Information, 22 can also be used in place of the Pearson correlation used here and give similar results.
This method successfully identified known therapeutic targets ERBB2 (HER2) and ESR1 in ER+ and HER2+ subtypes of breast cancer. These genes are already successfully targeted by hormonal therapy by compounds such as Herceptin (Genentech Inc.) for HER2+ or tamoxifen for luminal subtypes of breast cancers, respectively. Interestingly, it is known that tamoxifen treatment is less successful in the luminal B (LB) subtype compared to luminal A (LA). 8 Our gene centrality score suggests a possible explanation: the centrality score of ESR1 is 6.94 in LA and only 3.44 in LB, although ESR1 is equally overexpressed in both. This suggests that the reason tamoxifen does not work as well in LB as in LA may be that in LB, the tumor is not as dependent on ESR1 as in LA. In other words, the number of other genes affected by ESR1 in LB is much smaller than in LA, and hence, it is less susceptible to drugs like tamoxifen that function by blocking the estrogen pathway.
The therapeutic targets associated with breast cancer subtypes in Table 2 are either novel or currently part of clinical trials. Epidermal growth factor receptor EGFR was identified as having high centrality in high-risk ER+ tumors (luminal B), and our data would suggest that EGFR inhibitors should be targeted specifically at luminal B breast cancers. TGF-beta receptor 2 has very high centrality in luminal A tumors, suggesting that these tumors may benefit from TGF-beta inhibitors. Intriguingly, the ETS-related genes ERG (and ELK3 and ETS2) all had high centrality scores in luminal A tumors. ERG has been shown to be involved in translocations in a significant proportion of prostate cancer, 23,24 another hormone-sensitive solid tumor. Our data would suggest that ERG abnormalities may be present in the luminal A subclass of breast cancers as well.
PIM2 and a number of SRC tyrosine kinases including YES1 and LYN were predicted to be good therapeutic targets in subsets of basal-like and/or HER2+ breast tumors. LYN has recently been shown to be overexpressed in subsets of basal-like breast cancer and required for cell migration and invasion, but not for proliferation, in basal-like breast cancer cell lines. 25 The analysis from the present article suggests that YES1 is also a therapeutic target in basal-like breast cancer cell lines. Note that the SKBR3 cell line that profiles as “basal” in some studies but has HER2 amplification 42 was also sensitive to knockdown of YES1. These data suggest that, in addition to a subset of BLC, YES1 may also be a target in some HER2+ tumors. It also suggests that the thresholds we used for centrality scores may be too conservative and that genes with more modest elevations in centrality scores may also be therapeutic targets. Our study combined with Choi et al.’s 25 suggests that overexpression of YES1 and LYN could be used to guide treatments with SRC kinase inhibitors like Dasatinib (Bristol-Myers Squibb, New York, NY), AP 23846 (Ariad, Cambridge, MA), TG 100598 (TargeGen, San Diego, CA), AZD 0539 (AstraZeneca, London, UK), or SKI-606 (Wyeth, Madison, NJ). Dasatinib (Bristol-Myers Squibb) is a drug that inhibits the BCR/ABL pathway in addition to SCR kinases and has been shown to slow the growth of triple-negative (ER−/PR−/HER2−) breast cancer cell lines in vitro. 26,27 It is unclear whether this effect is due to the inhibition of BCR/ABL or any of the SRC kinases, but based on centrality scores and subsequent experiments, YES1 and LYN may be at least partially responsible for the phenotypic changes observed upon Dasatinib (Bristol-Myers Squibb) treatment.
Materials and Methods
Datasets and preprocessing
Previously published breast cancer microarray datasets (accession numbers GSE2034 14 and GSE4922 17 ) were obtained from the Gene Expression Omnibus website (GEO; www.ncbi.nlm.nih.gov/geo). The first dataset (GSE2034) comes from a study of Wang et al. 14 and consists of gene expression data from 286 lymph node–negative patients treated with surgery and radiation alone and followed for up to 150 months after treatment, with recorded events for distant metastasis (Suppl. Table S1). ER status was available, and HER2 status was known but not provided. The second dataset (GSE4922) from Ivshina et al. 17 consisted of 249 primary invasive breast tumors (Suppl. Table S2). In this cohort, 64% of patients were lymph node negative and were treated with surgery and radiation alone. The remaining set was lymph node positive and received systemic adjuvant polychemotherapy consisting of intravenous cyclophosphamide, methotrexate, and 5-fluorouracil. 28 Histological grade, tumor size, ER, and P53 biomarker information were available for each sample together with up to 153 months of follow-up information for distant metastasis.
Microarrays were MAS 5.0 normalized, and only probes present in both datasets were retained. Multiple probes corresponding to the same gene were compressed to the one with the biggest median over all arrays after taking log2 of each intensity value. In addition, every array was scaled to median zero by subtracting the median of each array from every expression value.
Robust, unsupervised consensus ensemble clustering methods previously applied to GSE2034 identified 6 core breast cancer subtypes 13,15,16 as listed in Supplementary Table S1: 2 ER+/HER2− subtypes: luminal A (LA) and luminal B (LB); 2 basal (ER−/HER2−) subtypes: BA1 and BA2; and 2 HER2+ subtypes: HER2I and HER2NI. The samples in GSE4922 were assigned a subtype as follows: HER2+ samples were identified based on 17q12 amplification using expression levels of ERBB2, GRB7, STARD3, and PPARBP. Gene expression values in both datasets were normalized by subtracting the median and dividing by the median absolute deviation. HER2+ samples were identified as those overexpressing ERBB2 and at least 2 others from the set GERB7, STARD3, and PPARBP. After HER2 samples were identified, the 2 datasets were merged using a method called Distance Weighted Discrimination (DWD), 29 which corrects for biases arising from different experimental conditions. The assignment of samples in Dalgin et al.’s study 15 to subtypes was done by comparison to mean expression profiles (centroids) across all genes for each subtype, using the classification of GSE2034 as the standard. This method, called Single Sample Predictor, 9 calculates a “distance” from each sample to mean expression values of samples in labeled sets using Euclidean distance or Pearson correlation and assigns them to the set for which this distance is minimum (Suppl. Table S2). Cases with inconsistent class labels for different distance metrics were discarded.
Meta-analysis of outliers
To minimize sample size bias, 10 arrays were randomly picked from each breast cancer subtype and combined into a reduced gene expression table,
The sampling procedure was repeated 1,000 times, separately for the 2 datasets (GSE2034 and GSE4922), and in each sampling, outlier scores were generated for each gene in each subtype. At the end of this analysis, every gene had 2 associated distributions of outlier scores for each subtype that could now be combined into a single consensus score. This metaoutlier score was calculated, using the method of Cochran, 31 as a weighted mean of the average outlier scores from the 2 distributions (θ̄ 1 and θ̄ 2), where the weights were the inverse of corresponding variances σk 2:
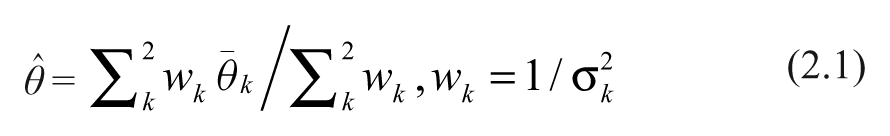
Each gene was now assigned a metaoutlier score for each of the 6 breast cancer classes (BA1, BA2, HER2I, HER2NI, LA, and LB), which estimates whether it is relatively overexpressed or underexpressed with respect to other subtypes.
Meta-analysis of correlations
Pearson correlations were computed between all pairs of genes within each subtype. Assuming a common underlying population correlation between every 2 genes in each class, we calculated metacorrelation values by first transforming each Pearson correlation r with a Fisher z transform:
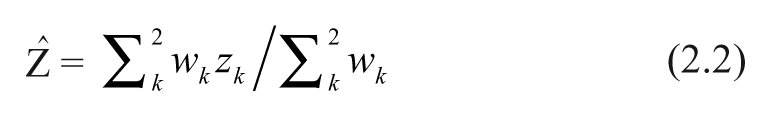
where z
1 and z
2 are z-transformed Pearson correlations between any 2 genes from datasets GSE2034 and GSE4922, respectively. The weights are w
k = n
k − 3, where n
1 and n
2 are numbers of samples used to calculate correlations in the 2 datasets. Since correlation values calculated from gene expression arrays are often noisy,
33
a homogeneity χ2 statistic,
Metacorrelation values were calculated using the inverseFisher z transform:
Gene centrality
Eigenvector centrality 35,36 is a measure of the importance of a node in a network. Relative scores are assigned to each node based on the idea that connections to nodes with high scores should contribute more to the score of the node in question than equal connections to low scoring nodes. Similarly, gene centrality is a measure of the importance of a gene in a modified gene network, where directed edges between nodes (genes) are weighted by a positive measure of the overexpression of the target gene as shown in the toy gene network from Figure 3. More generally, connections between nodes can be real positive numbers representing connection strengths.
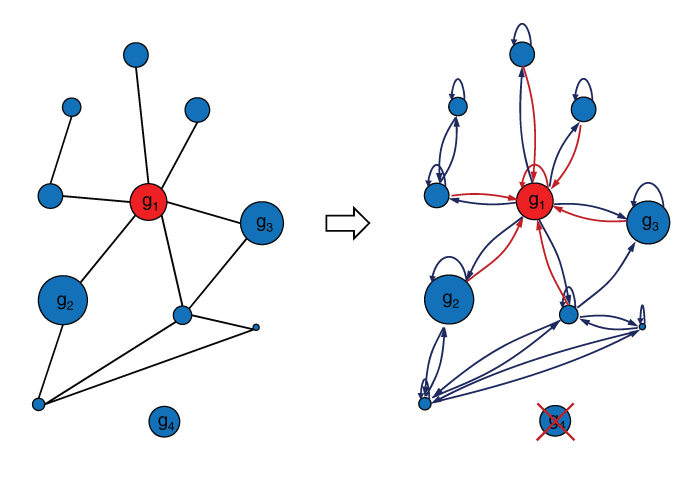
The gene centrality principle. Cartoon showing a toy gene network, with relative expression levels g i proportional to node size, transformed into a similar network with a primitive adjacency matrix. This is accomplished by eliminated unconnected genes and by adding self-loops to all nodes. In addition, undirected edges are changed into directed edges with associated weights equal to the expression level of the target gene. In this configuration, the center node (gene), which is colored red, would have the highest centrality score because of its high expression and connectivity.
Let
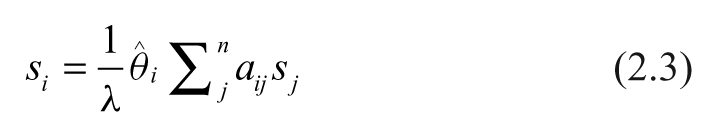
Here, λ is a constant of proportionality to be determined. Let

Equation 2.4 identifies λ as an eigenvalue of the product of matrices

where
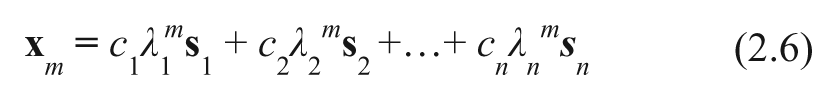
The linear system defined in equation 2.5 is completely characterized by the matrix
If
A primitive matrix
Separate
Identifying candidate therapeutic targets
Outlier scores (θ) and Pearson correlation values (r) were calculated for each gene across all samples. The outlier score is a measure of the relative overexpression of a gene in one subtype compared to all others. It is defined as the percentage of tumor samples that overexpress a particular gene in the subtype. To make the score robust, the outlier score for each subtype was defined as the mean over the distribution of outlier scores across bootstrap samples. To reduce sample size bias, each bootstrap dataset was chosen by random sampling of an equal number of samples from each subtype. Outlier score values were determined separately for the 2 datasets (GSE2034 and GSE4992) and then merged into one metaoutlier score by taking a weighted mean of the individual scores over bootstrap datasets. Similarly, Pearson correlation values between gene pairs were calculated for each of the 6 tumor classes for both datasets and then merged into metacorrelation values. Correlations that were significantly different between the 2 datasets were discarded. Centrality scores for each gene were calculated for the networks built from Pearson correlations between pairs of genes and outlier scores, using a procedure described in the Materials and Methods section. To allow comparison of scores across subtypes, centralities within each subtype were sorted and then divided by the median value over the entire gene set.
High correlation scores are transitive (linked across several genes) and can identify cliques of overexpressed genes with similar centrality scores. To find the genes most likely to cause a phenotypic change upon knockdown, we pruned the genes with high centrality scores in each subtype to known oncogenes. These were obtained from Agilent (Santa Clara, CA) and came as part of the GeneSpring bioinformatics software package. Oncogenes with high centrality scores identified by our analysis are presented in Table 2 along with the associated outlier scores calculated by meta-analysis over GSE2034 and GSE4992 gene expression tables. The full list of genes from the combined analysis is given in Supplementary Table S3, while Supplementary Tables S4 and S5 list the results for the individual analysis of GSE2034 and GSE4922.
Immunohistochemistry
Anti-YES1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was first optimized on human breast tissue microarray slides using the Discovery XT (Ventana Medical Systems, Tucson, AZ) automated immunostainer. Before hybridization, breast cancer tissue slides were deparaffinized in a 60°C oven for 1 hour followed by 3 × 5 minutes in xylene, and hydrated in 100%, 80%, and 70% ethanol and dH2O. Antigen retrieval was performed by using Cell Conditioning Solution (Ventana Medical Systems) for 72 minutes. Anti-c-Yes antibody was applied at a dilution of 1:30 and incubated at 37°C for 1 hour, followed by 12 minutes with a universal secondary antibody (Ventana Medical Systems). DABMap (Ventana Medical Systems) was used for chromogenic detection after which slides were counterstained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI) and dehydrated in 70%, 80%, and 100% ethanol.
Cell culture conditions
MDA468 and MDA231 cells were maintained in DMEM/F12 (Gibco, Grand Island, NY) supplemented with 5% fetal bovine serum (FBS) (Gibco), 1% amino acid (Cellgro, Manassas, VA), and 1% sodium pyruvate (Sigma, St. Louis, MO); BT549 and SKBR3 cells were maintained in RPMI 1640 (ATCC) with 10% FBS; MCF7 and HEK-293T in DMEM (Gibco) with 10% FBS and MCF10A were grown in DMEM/F12 to which the following were added: 5% horse serum (Invitrogen, Carlsbad, CA), 20 ng/mL epidermal growth factor (Invitrogen), 100 ng/mL cholera toxin (Sigma), 0.01 mg/mL insulin (Sigma), and 500 ng/mL hydrocortisone (Sigma). With the exception of HEK-293T cell culture media, all presented solutions had an addition of 1% penicillin/streptomycin (Gibco).
Immunoblotting
After incubation, cells were washed in cold (4°C) PBS solution and then kept on ice with NETN buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.5% NP40, 1x protease inhibitor cocktail [Sigma]) for 15 minutes. Cells were then scraped and collected in 1.5-mL tubes and incubated on ice for 5 minutes. Whole cell protein was then extracted by sonication followed by 14,000 rpm centrifugation for 10 minutes. The supernatant was then collected and quantified by using a Bradford-based 38 protein assay (Bio-Rad, Hercules, CA). After loading 25 to 50 µg of protein onto 10% polyacrylamide gels, they were subject to electrophoresis, transferred to PVDF membranes (Bio-Rad), and probed with antibodies against YES1 (1:1,000; BD Transduction Laboratories, San Diego, CA) and GAPDH (1:5,000; Abcam, Cambridge, UK).
Lentivirus production
To suppress YES1, we introduced shRNA specific for the following sequences using pLKO.1 lentiviral vectors 39 acquired from Open Biosystems: shYES1 #1: CCAGCCTACATTCACTTCTAA; shYES1 #2: ACCACGAAAGTAGCAATCAAA; and shYES1 #3: CCTCGAGAATCTTTGCGACTA. A standard 18-bp nonhairpin control (CCGCAGGTATGCACGCGT) was also acquired from Addgene (Cambridge, MA) together with psPAX2 packaging plasmid and pMD2.G envelope plasmid. Lentiviruses were produced by transiently transfecting individual shRNA constructs together with packaging and envelope plasmids into HEK-293T cells using Fugene 6 (Roche, Basel, Switzerland). Viral supernatants were collected and passed through 0.45-µm syringe filters.
Cell proliferation assays
Cells were plated in 6-cm culture dishes and grown in the incubator until they were 70% confluent. After changing to fresh culture media, 8 µg/mL of polybrane (Millipore, Billerica, MA) was added together with 0.5 mL of each of the previously prepared lentiviral solutions to separate dishes: one for the lentivirus containing the scrambled shRNA (shSRC) and one corresponding to the lentivirus designed to knock down the expression of YES1 (shYES1). After 24 hours, the media containing viral particles were replaced with fresh media to which 3 µg/mL puromycin (Sigma) was added in order to select for infected cells. The cells kept on growing for 3 to 4 days until a stable population was obtained.
Cells expressing shYES1 and shSCR were separately plated in triplicates in 12-well plates in the following quantities: 50 × 103 cells for MDA231, BT549, and MCF10A; 25 × 103 cells for MDA468 and SKBR3; and 10 × 103 cells for MCF7. After 6 days of growing in specific media supplemented by 3 µg/mL puromycin, cells in each well were collected and counted by trypan blue exclusion using a Beckman Coulter counter (Brea, CA).
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported in part by the New Jersey Commission on Cancer Research (to E.B.), grant number: 09-112-CCR-E0.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.