
Abstract
Epigenetic modifications are heritable changes in gene expression not encoded by the DNA sequence. In the past decade, great strides have been made in characterizing epigenetic changes during normal development and in disease states like cancer. However, the epigenetic landscape has grown increasingly complicated, encompassing DNA methylation, the histone code, noncoding RNA, and nucleosome positioning, along with DNA sequence. As a stable repressive mark, DNA methylation, catalyzed by the DNA methyltransferases (DNMTs), is regarded as a key player in epigenetic silencing of transcription. DNA methylation may coordinately regulate the chromatin status via the interaction of DNMTs with other modifications and with components of the machinery mediating those marks. In this review, we will comprehensively examine the current understanding of the connections between DNA methylation and other epigenetic marks and discuss molecular mechanisms of transcriptional repression in development and in carcinogenesis.
Introduction
DNA methylation is a heritable epigenetic mark involving the covalent transfer of a methyl group to the C-5 position of the cytosine ring of DNA by DNA methyltransferases (DNMTs). 1 In plants, cytosines are methylated in both symmetrical (CG or CHG) or asymmetrical (CHH, where H is A, T, or C) contexts. In mammals, DNA methylation occurs at cytosines in any context of the genome. 2 However, more than 98% of DNA methylation occurs in a CpG dinucleotide context in somatic cells, while as much as a quarter of all methylation appears in a non-CpG context in embryonic stem cells (ESCs). 2 DNA methylation is typically removed during zygote formation and then re-established in the embryo at approximately the time of implantation. 3 Most DNA methylation is essential for normal development, and it plays a very important role in a number of key processes including genomic imprinting, X-chromosome inactivation, and suppression of repetitive element transcription and transposition and, when dysregulated, contributes to diseases like cancer.1,4-6
DNA methylation is regulated by a family of DNMTs: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L.7-11 DNMT1 preferentially methylates hemimethylated DNA in vitro and is localized to replication foci during S phase. As such, it is the proposed maintenance methyltransferase responsible for copying DNA methylation patterns to the daughter strands during DNA replication. 12 Mouse models with both alleles of Dnmt1 deleted are embryonic lethal at approximately day E9.13,14 DNMT2 is a methyltransferase homolog that methylates cytosine-38 in the anticodon loop of aspartic acid transfer RNA instead of DNA. 15 DNMT3A and DNMT3B, in contrast to DNMT1, have preference for unmethylated CpG dinucleotides and perform de novo methylation during development. Mice lacking Dnmt3a die at about 4 weeks of age, whereas Dnmt3b knockout induces embryonic lethality at E14.5 to E18.5.13,16 Possessing homology to DNMT3A and DNMT3B, DNMT3L assists the de novo methyltransferases by increasing their ability to bind to the methyl group donor, S-adenosyl-L-methionine (SAM), and stimulating their activity in vivo, 17 although DNMT3L has no catalytic activity itself. Dnmt3L homozygous-null mice are viable, whereas heterozygous embryos derived from homozygous Dnmt3L-null oocytes die around E9 and display impaired maternal methylation imprints and biallelic expression of imprinted genes normally expressed only from the allele of paternal origin. 18 Cooperation among different DNMTs is also required in methylating some regions of the genome, particularly repetitive elements. As previously mentioned, it has been widely believed that DNMT1 acts mainly as a “maintenance” methyltransferase during DNA synthesis and that DNMT3A and DNMT3B act as “de novo” enzymes in development. However, mounting evidence indicates that DNMT1 may also be required for de novo methylation of genomic DNA 19 and that DNMT3A and DNMT3B contribute to maintenance methylation during replication. 20
In mammals, most CG dinucleotides are methylated on cytosine residues, whereas CG dinucleotides within promoters tend to be protected from methylation. Defects in DNA methylation are closely associated with cancer, although no mutation or deficiency in any DNMT has been identified as causally linked to tumor development, most likely because of their critical role during embryogenesis. Epigenetic hallmarks of cancer include global DNA hypomethylation and locus-specific hypermethylation of CpG islands (CGIs). 21 Thus far, all examined tumor samples display global reductions of DNA methylation. 22 Hypomethylation arises mainly from loss of methylation at normally heavily methylated repeat elements, including satellites (e.g., SAT2) and retrotransposons (e.g., LINEs), leading to genomic instability and oncogene activation. Locus-specific hypermethylation usually occurs at promoter CGIs of tumor suppressor genes, resulting in heritable transcriptional silencing. Methylation of DNA may itself physically impede the binding of transcriptional regulators to the gene, 23 and more importantly, methylated DNA participates in the formation of chromatin through interactions with various other epigenetic modifications such as the histone code, polycomb complexes, nucleosome positioning, noncoding RNA, and ATP-dependent chromatin remodeling proteins. 21 Therefore, we next examine the links between DNA methylation and other epigenetic regulators to better understand the molecular mechanisms of transcriptional repression in development and carcinogenesis.
Links between DNA Methylation and Histone Modifications
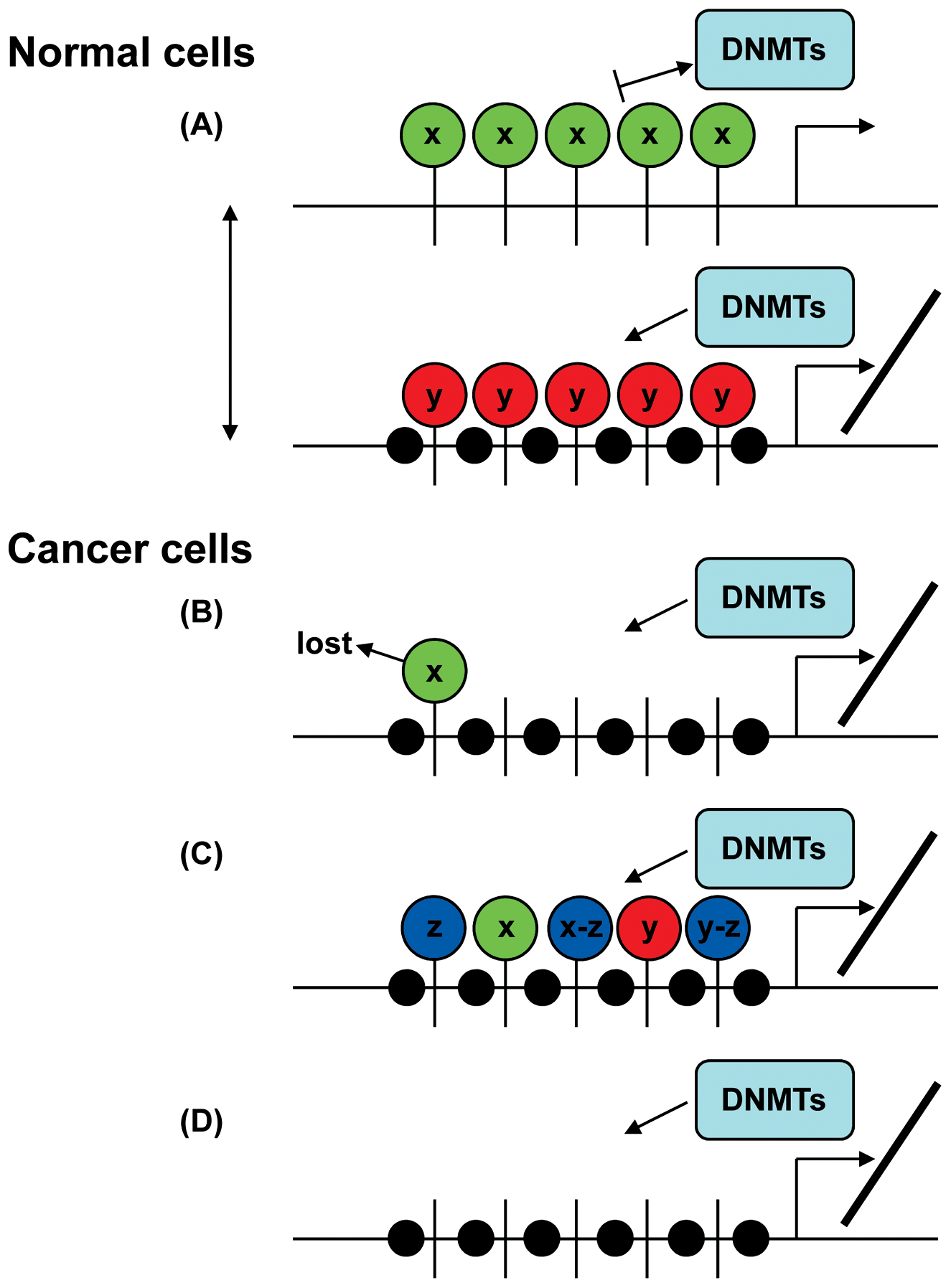
Covalent modification of DNA and histone proteins, the core components of chromatin, provides a heritable mechanism for regulating gene expression. Histone tails undergo a variety of covalent modifications including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation, regulating key cellular process such as gene transcription, DNA replication, and DNA repair.24,25 During mammalian development and cancer, DNA methylation and specific histone modifications appear to reciprocally influence each other in deposition: histone methylation may direct DNA methylation patterns (Fig. 1), and DNA methylation may serve as a template for the establishment of certain histone modifications (Fig. 2) after DNA replication. 24
How the histone code may direct DNA methylation during development and carcinogenesis. (
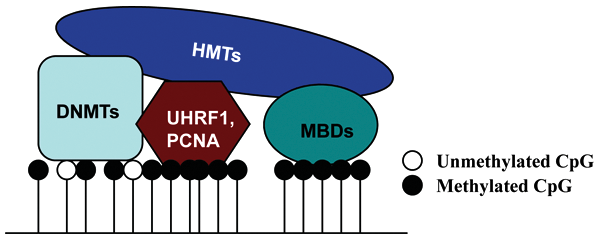
How the histone code may rely on the DNA methylation machinery for direction. Upon binding to CpG-rich regions, DNMTs may directly recruit HMTs to these domains. During DNA replication, UHRF1 preferentially binds hemimethylated DNA and interacts with/recruits DNMT1 and G9A. PCNA may also have a role in the recruitment process. Methyl-CpG–binding proteins (MBDs) specifically interact with methylated DNA and may form complexes with HMTs such as SETDB1 to direct histone methylation to regions of DNA methylation.
Trimethylation of histone H3 lysine 9 (H3K9), histone H3 lysine 27 (H3K27), and histone H4 lysine 20 (H4K20) has been suggested to be a prerequisite for subsequent DNA methylation, which appears to be attributable to physical associations between the components of these histone methylation systems and one or more DNMTs (Fig. 1). In the context of H3K9 and H3K27, the relevant histone lysine methyltransferases, SUV39H1/2 and EZH2, respectively, interact directly with DNMT1, DNMT3A, and DNMT3B.26,27 Pericentromeric localization of DNMT3B depends on SUV39H1/ 2-mediated H3K9 dimethylation or trimethylation. Suv39h1/2 double-null murine embryonic stem cells display reduced DNA methylation levels at major satellite repeats but not at minor satellites or endogenous C-type retroviral elements. 26 Endogenous DNMT1 and DNMT3A also associate with H3K9 methyltransferase SUV39H1 activity as well, mediated by the conserved PHD-like motif in the case of DNMT3A. 28 EZH2, one of the core components of polycomb repressive complex 2 (PRC2), catalyzes trimethylation of H3K27, which acts subsequently to recruit the PRC1 complex and mediate transcription silencing. EZH2 physically interacts with the DNMTs and facilitates their binding to certain EZH2 target promoters. 27 Overexpression of EZH2 increases CpG methylation, while RNA interference knockdown of EZH2 reduces H3K27 methylation and DNA methylation at known EZH2 target genes. 27 G9A, another histone methylase, catalyzes monomethylation and dimethylation of H3K929,30 and, to a lesser extent, H3K27. 29 Genome-wide DNA methylation analysis of G9a knockout murine cells reveals that it site-selectively contributes to DNA methylation. Reduction of DNA methylation occurred at only 1.6% of the approximately 2,000 genic loci analyzed in the G9a knockout ESCs. 31 The G9A/GLP heteromeric complex induces both H3K9 and DNA methylation on G9A target loci, and these two epigenetic marks cooperatively suppress transcription of their target genes. Tachibana et al. showed that promoter regions of G9A/GLP target genes are DNA hypomethylated in G9a or Glp knockout ESCs. 32 Histone arginine methylation has also been linked to DNA methylation and gene silencing. 33 Symmetric dimethylation of histone H4 arginine 3 (H4R3me2s), catalyzed by PRMT5, serves as a direct binding target for DNMT3A, 33 which may then induce methylation of adjacent CpG dinucleotides at PRMT5 target genes. In addition to the direct recruitment of DNMTs, histone methyltransferases and demethylases may also influence the stability of DNMT proteins.34,35 SET7, a known histone methyltransferase for H3K4, colocalizes with DNMT1 and methylates DNMT1 at the K142 position to regulate its stability and degradation. 34 Overexpression of SET7 leads to decreased DNMT1 levels, and siRNA-mediated knockdown of SET7 stabilizes DNMT1. Lysine-specific demethylase 1 (LSD1) has been shown to demethylate histone H3K4 and H3K9. LSD1 also demethylates DNMT1 methylated at K142 to antagonize the effect of SET7. 35 Therefore, Lsd1 deletion in murine ESCs induces progressive loss of DNA methylation.
HP1 (heterochromatin protein 1) proteins also provide a link between DNA methylation and histone marks. HP1 binding protein HP1-BP74 directly binds to HP1, and its central domain associates with linker DNA at the entry/exit site of the nucleosome in vitro. 36 HP1 proteins modulate gene transcription in both euchromatin and heterochromatin. HP1 binds to methylated H3K9 through its N-terminal chromodomain. The binding of HP1 to constitutive heterochromatin depends on the enzymatic activity of SUV39H1/2, which catalyze trimethylation of H3 at lysine 9, 37 while HP1 binding to euchromatin depends on dimethylation of H3K9 mediated by G9A. 30 Methylated H3K9 serves as a binding platform for HP1 to associate with the DNA methylation machinery. HP1 binds directly to the PHD-like motif of DNMT3A in vitro. 28 The direct physical link identified between the DNMTs and the H3K9me-HP1 system therefore ensures that H3K9 methylation directly influences DNA methylation patterns.26,28 Smallwood et al. 38 reported that DNMT1 also interacts with HP1, leading to increased methylation on DNA and chromatin templates in vitro. The functional and physical interactions were recapitulated in vivo as well. Binding of GAL4-HP1 to a reporter construct is sufficient to induce repression and DNA methylation in DNMT1 wild-type but not DNMT1-null cells. Additionally, silencing of the survivin gene coincides with recruitment of G9A and HP1 in DNMT1 wild-type but not DNMT1-null cells. Therefore, direct interactions between HP1 and DNMT1 mediate silencing of euchromatic genes. HP1 proteins also recruit a variety of other factors including histone deacetylases, transcriptional repressors, and chromatin remodeling enzymes to the methylated region to further enhance and/or stabilize repressive domains.1,39
In addition to the previously discussed links, certain components of the histone code may utilize or rely on the DNA methylation machinery as a template for their deposition (Fig. 2). DNA methylation is faithfully reproduced through semiconservative replication to copy the heritable information from a template. However, there is no obvious template for nucleosome reassembly after replication. PCNA (proliferating cell nuclear antigen), present at replication forks, plays a very important role both in DNA synthesis, as the polymerase processivity factor, and in the inheritance of epigenetic marks, which may act as a guide for other epigenetic modifications. 12 PCNA recruits a variety of epigenetic regulators such as the histone modifiers HDACs and SETD8, chromatin remodeler SMARCA5, and chromatin assembly factor 1 (CAF1). 12 The maintenance of DNA methylation at the replication fork is likely ensured by the DNMT1-PCNA and/or DNMT1-UHRF1 interactions. While disruption of the DNMT1-PCNA interaction does little to alter genomic DNA methylation levels, disruption of the DNMT1-UHRF1 interaction results in massive genomic hypomethylation, 40 suggesting that UHRF1 is the key player coordinating with DNMT1 to maintain DNA methylation patterns after DNA replication. UHRF1 preferentially binds to hemimethylated DNA, interacts with DNMT1, and is required for DNMT1 localization to replicating heterochromatic regions.41,42 In addition, UHRF1 specifically interacts with peptides that are methylated at H3K9 in vitro,12,43 and it resides in a complex with HDACs and G9A. 41 Therefore, in addition to binding hemimethylated DNA, UHRF1 appears to interpret the local histone environment, thereby creating a feedback mechanism that involves the mutual reinforcement of histone and DNA methylation marks.
There is also evidence that DNA methylation directs H3K9 dimethylation or trimethylation, although this remains controversial. Hypomorphic mutation of DNMT1 gene causes a global reduction of H3K9me2 and H3K9me3 levels in a cancer cell line; re-expression of DNMT1 rescues this phenotype.44,45 Dnmt1 and Dnmt3b double knockout in colon cancer cells induces changes in H3K9 methylation levels at heterochromatin and specific tumor suppressor loci. 46 Treatment of breast, bladder, and colorectal cancer cell lines with the demethylating agent 5-aza-2’-deoxycytidine (5-aza-CdR) results in reactivation of multiple aberrantly silenced genes and a concomitant decrease in H3K9 methylation. 47 Triple knockout (TKO) of Dnmt1/Dnmt3a/Dnmt3b in ESCs results in complete loss of DNA methylation at several repetitive sequences and imprinted genes 48 ; however, TKO ES cells retain occupancy of H3K9 methylation and HP-1 in pericentromeric regions. 48 The link between DNA methylation and H3K9 methylation is via interaction of the enzymes mediating these marks. DNMT1, for example, directly binds G9A both in vivo and in vitro, and these two proteins colocalize in the nucleus during DNA replication. A complex of DNMT1 and G9A colocalizes with H3K9me2 at replication foci. Depletion of DNMT1 not only impairs DNA methylation but also retards G9A loading and H3K9 methylation on chromatin and rDNA repeats. 44 Similarly, heterochromatic colocalization of SUV39H1 and DNMT1 exists exclusively before cell division.
Yet an additional link between DNA methylation and the histone code exists via the methyl-CpG–binding proteins (MBDs), which interact specifically with methylated DNA and mediate transcriptional repression (Fig. 2). During replication of DNA methylation-rich regions of the genome, CAF1 forms a complex with MBD1 and the histone lysine methyltransferase SETDB1, thereby coupling histone methylation with DNA methylation. 12 Mbd1 deletion analysis and coimmunoprecipitation experiments thus far suggest that H3K9 methylation mediated by SETDB1 is dependent on MBD1, which recruits SETDB1 to CAF-1 at active replication forks. 49 The two global mechanisms of gene regulation, DNA methylation and histone deacetylation, are also linked via the methyl-CpG– binding protein MeCP2. MeCP2 binds tightly to chromosomes in a DNA methylation–dependent manner. It contains a transcriptional-repression domain (TRD) that functions at a distance in vitro and in vivo, and MeCP2 associates with a corepressor complex containing histone deacetylases. 50
An emerging and potentially quite significant finding from genome-wide studies is the dramatic inverse correlation between DNA methylation and H3K4 methylation (Fig. 1). A recent genome-wide analysis revealed that the conserved DNA methylation of CGIs inversely correlates with the presence of trimethylation of H3K4, 51 although the mechanism is still far from clear. CXXC finger protein 1 (CFP1), encoded by the CXXC1 gene, is essential for mammalian development and is an important regulator of chromatin structure. CFP1 selectively binds to unmethylated CpGs in vitro 52 and in vivo. 53 High-throughput next-generation sequencing of Cfp1-bound chromatin identified a notable concordance between unmethylated CGIs and sites of H3K4me3 in the mouse brain. 53 Levels of H3K4me3 at CGIs are markedly reduced in Cfp1-depleted cells, consistent with the finding that CFP1 associates with the H3K4 methyltransferase SETD1. DNA methylation–free CpG clusters recruit CFP1, and probably other CXXC domain– containing proteins as well, to stimulate methylation of H3K4. Densely methylated CGIs, on the other hand, attract methyl-CpG–binding proteins, which in turn recruit enzymes that reinforce repressive histone marks. Peptide interaction assays revealed that DNMT3L specifically interacts with the extreme amino terminus of histone H3; this interaction, however, was strongly inhibited by methylation of H3K4 but was insensitive to modifications at other positions. DNMT3L recognizes histone H3 tails that are unmethylated at lysine 4 and may therefore induce de novo DNA methylation by recruitment and activation of DNMT3A2. 54 In mammals, there are at least 10 known or predicted H3K4 methyltransferases, which are generally categorized into the MLL (mixed lineage leukemia) family, SET1 family, and others. 55 H3K4 methylation may protect certain loci from DNA methylation. Disruption of the SET domain of MLL reduced H3K4me1 levels and increased DNA methylation levels at specific loci (e.g., Hoxd4) but not globally in a mouse model. 56 However, forced overexpression of exogenous Mll in Mll knockout cells did not reduce global DNA methylation levels. 57 It is therefore possible that DNA methylation represents a default state of the genome unless H3K4me, or possibly other histone marks, is present to maintain specific regions (e.g., promoters) free of DNA methylation to permit proper gene regulation (Fig. 2).
Polycomb Group (PcG) Proteins and DNA Methylation in Development and Cancer
PcG proteins were first discovered in Drosophila melanogaster as key regulators of homeotic (Hox) gene expression, which is critical for normal body patterning. In mammals, two main protein complexes, PRC1 and PRC2, have been identified, and both play fundamental roles in transcription silencing. The mammalian PRC1 complex is diverse and includes HPH (1, 2, and 3), RING1/RING2, RYBP, and BMI1 (or its homologs MEL18 and NSPC1) as core subunits. PRC2 has three core components EZH2, EED, and SUZ12. 58 Trimethylation of H3K27, catalyzed by the PRC2 subunit EZH2, is a central feature of PcG-silenced chromatin that provides a “docking site” to recruit the PRC1 complex.58-61 PRC1 is a direct executor of silencing at target genes. PRC1 complexes have at least two central functions: the first (originally defined for PRC1) is compaction of chromatin, and the second (defined with the BCL6 corepressor, or BCOR, complex) is catalysis of histone H2A monoubiquitination.62,63 Unlike DNA methylation, polycomb-mediated repression is considered readily reversible. Lineage-specific transcription factors and HOX gene clusters are the primary targets of H3K27me3 in ESCs. Most of these loci are transcriptionally active upon terminal differentiation.64-66 It is currently believed that repression by polycomb proteins is established early in development then lost in a lineage- specific manner upon differentiation. 65 However, a subset of promoters marked by H3K27me3 in stem cells frequently acquire DNA methylation during differentiation, which adds another layer of repression to stably, and perhaps permanently, lock the selected genes in the “off” state, suggesting that context-dependent crosstalk between polycomb and DNA methylation exists, although much remains unknown about this interaction. 65
PcG complexes, like the DNMTs, have strong links to cancer. For example, EZH2 is overexpressed in tumors and is predictive of poor prognosis. 67 BMI1 cooperates with MYC to promote lymphomas by repressing the INK4a/ARF tumor suppressor locus. Genes targeted by polycomb complexes are generally associated with CGI promoters and, as such, are protected from de novo methylation at the time of implantation. Thus, most EZH2 target genes actually remain constitutively unmethylated throughout development. 66 Nonetheless, a number of these genes might become targets for de novo DNA methylation under pathological conditions and contribute to cancer. Connections between DNA methylation and PcG have continued to accumulate. Schlesinger et al. showed that genes subject to tumor-specific hypermethylation in colon cancer were more likely to be marked by H3K27 methylation in normal tissues than genes lacking H3K27 methylation. 68 Widschwendter et al. reported that PcG targets in ESCs were 12-fold more likely to become methylated in cancer. 69 Nearly 49% of genes methylated in colon cancer were PcG targets in ESCs in another study. 70 Interestingly, PcG targets in stem cells are far more likely to become methylated than nontargets with age, 71 the most important demographic risk factor for cancer. DNA methylation of PcG target genes is present in preneoplastic conditions and may lead to aberrant gene expression associated with carcinogenesis. 71 Our laboratory reported that approximately 47% of DNMT3B-regulated genes were bound by PRC1 or PRC2 in human colon cancer, and we demonstrated a novel link between DNMT3B and the mark mediated by PRC1: knockout of DNMT3B resulted in loss of Ub-H2A at several PRC1 target genes. 5 Our results are supported by the findings of Kallin et al., who discovered that H2A ubiquitination was enriched at high-density CpG promoters, suggesting that DNA methylation may be linked to uH2A at these regions. 72 In contrast, Gal-Yam et al. reported that many genes hypermethylated in a prostate cancer cell line were bound by PcG in normal cells but lost PcG binding upon acquisition of DNA methylation in the context of prostate cancer cell lines. 73 These studies demonstrate that compelling connections between DNA methylation and PcG proteins exist (Fig. 2). Yet, exactly how PcG influences or recruits DNA methylation remains uncertain.
Most polycomb target promoters exist in a bivalent configuration in ESCs, marked by both repressive H3K27me3 and activating H3K4me3 marks. 66 However, about 40% of ESC bivalent domains are preserved upon differentiation, indicating that most genes harboring bivalent modifications are driven into either an active or an inactive state.66,74 Thus, genes that are silenced by this mechanism maintain the possibility of being readily activated, whereas genes in their active conformation might easily revert to the repressed state upon differentiation. Differentiated cells lose bivalent modifications at many genes and acquire a more stable, less plastic, chromatin structure to maintain cell fate during cellular expansion, which is supported by recent findings that repressive marks like DNA methylation or H3K9 methylation are added to “lock” in cellular states upon differentiation.65,75 Embryonic carcinoma cells are pluripotent and possess a stem cell–like chromatin configuration with bivalency predisposing to hypermethylation of tumor suppressor genes,76,77 supporting the “cancer stem cell” hypothesis that adult cancers may derive from stem cell–like cells, early progenitor cells, or dedifferentiated somatic cells. This theory is based on a number of observations: tumors harbor rare cells expressing stem/precursor markers, tumors are heterogeneous with only a subset of cells possessing tumor regeneration capacity, and many pathways important to the maintenance of stem/precursor cells are upregulated, mutated, or constitutively activated in tumor cells.77,78
Cancer has both a genetic and epigenetic basis. Clonal genetic changes are common in tumors, and characteristic genetic changes clearly contribute to the development of leukemia and solid tumors. However, genetic alterations are not the only mechanism that leads to dysregulated gene expression patterns during carcinogenesis. Epigenetic alterations are ubiquitous and an alternative to genetic changes such as mutations and transposition in gene disruption. 79 The cancer stem cell model suggests that epigenetic changes, which occur in normal stem or early progenitor cells, are the earliest events in cancer initiation. This view is strongly supported by the finding that DNA methylation–induced silencing of tumor suppressor genes occurs in the earliest stages of tumorigenesis. Early aberrant epigenetic changes may occur in normal cells under stress conditions such as chronic inflammation, well before tumors arise.22,80 Feinberg et al. 22 suggested that disruption of epigenetic marks in progenitor cells is a key determinant not only of cancer risk but also of tumor progression and tumor heterogeneity. In embryonic stem cells, some genes are held in a “transcription-ready” state mediated by the bivalent promoter chromatin state. The repressive chromatin modifications in cancer cells resemble those observed in normal embryonic stem cells, which may help prevent stem/precursor cells from committing to differentiation until programmed to do so. 77 The bivalent chromatin pattern may render groups of genes more vulnerable to errors that result in recruitment of aberrant promoter DNA methylation early during the progression of adult cancers, abnormally locking in the silenced state of genes (particularly prodifferentiation genes) and making them incapable of responding properly to differentiation cues. For example, embryonic carcinoma cells add two key repressive marks to bivalent genes, H3K9me2 and H3K9me3, which are both associated with aberrantly DNA hypermethylated genes in adult cancers. Hypermethylation of select promoters may be mediated by EZH2 since DNMT1, DNMT3A, and DNMT3B interact with EZH2 and EZH2 targets DNA methylation to certain promoters in vitro. 27 However, it must be noted that the presence of histone methylation at H3K27 mediated through EZH2 does not always induce DNA methylation 69 ; so, it is clear that other factors must be involved in triggering this cell type–specific de novo methylation, and it is these mechanisms that need to be further clarified.
Nucleosome Positioning and DNA Methylation
Core histones H2A, H2B, H3, and H4 form nucleosome particles that package approximately 147 bp of DNA, and the linker-histone H1 packages additional DNA between core particles, forming chromatin. Chromatin, rather than naked DNA, is the substrate for all processes that affect genes and chromosomes. Nucleosomes form the fundamental repeating unit of eukaryotic chromatin, serving as the basic module for DNA packaging that regulates the accessibility of DNA to interacting proteins and alters transcriptional states. 81 Recent studies have revealed that nucleosomal DNA is an important substrate for the DNMTs in vivo.82,83 While recombinant DNMT1 and DNMT3 enzymes are able to methylate CpG sites on nucleosomes assembled in vitro, few studies have addressed how they interact with chromatin in vivo. Jeong et al. 82 reported that both DNMT3A and DNMT3B are strongly anchored to nucleosomes, whereas DNMT1 interacts primarily with linker DNA. DNMT3L binding to unmethylated H3K4 tails clearly provides a link between DNMT3L/DNMT3A and nucleosomes in ESCs 54 ; however, DNMT3L is expressed only during gametogenesis and embryonic stages, 18 suggesting that other mechanisms are required for directing DNMTs to specific chromatin regions in somatic cells. When histone-DNA interactions within the nucleosome are disrupted by the DNA intercalator ethidium bromide, DNMT3A and DNMT3B dissociate from the histone proteins and cosediment with free DNA, suggesting that the enzymes depend on the interaction with DNA, as well as with histones, for stable anchoring. Deletion experiments demonstrated that the N-terminal region of DNMT3A and DNMT3B plays an essential role in their strong nucleosomal binding. 82 DNMT3A and DNMT3B contain PWWP- and PHD-like motifs in their N-terminal regulatory domains. DNMT3A was recently shown to specifically bind the H3K36me3 mark through its PWWP domain, which is important for the subnuclear localization of DNMT3A and for its catalytic activity on native chromatin. 84 This result suggests that nucleosomal binding of DNMT3A and DNMT3B might, at least in part, be mediated by its interaction with H3K36me3. Surprisingly, some well-known chromatin-modifying proteins, such as PCNA, HP1, MBD2, EZH2, HDAC1, and UHRF1, are not required for DNMT3A and DNMT3B to bind to nucleosomes. 82 Methylated SINE and LINE repetitive elements and CGIs in nucleosomes represent the main binding sites of DNMT3A and DNMT3B, suggesting that additional cues, other than hemimethylated DNA, from the chromatin are needed for inheritance of DNA methylation.
Nucleosome positioning also has a striking effect on DNA methylation. As a linker histone, H1 reduction leads to decreases in nucleosome repeat length, 85 which is a primary determinant of nucleosome spacing. Interestingly, depletion of histone H1 induces DNA hypomethylation of specific CGIs such as the imprinting control regions of the H19-Igf2 and Gtl2-Dlk1 loci, 85 indicating that linker histones participate in epigenetic regulation of gene expression by contributing to the maintenance and/or establishment of specific DNA methylation patterns. The mechanism by which H1 accomplishes this, however, remains unknown. It is well known that the composition of the nucleosome core particle influences chromatin structure and nucleosome positioning. Recent emerging evidence has revealed that replacement of the core histones with variants of histone H2A or H3 represents another potential means of gene regulation.86-89 The histone H2A variant H2A.Z is excluded from regions of methylated DNA, and H2A.Z enrichment varies inversely with transcription within gene bodies, a finding conserved from plants to animals, 89 indicating that H2A.Z incorporation may contribute to transcriptional activation by protecting genes against DNA methylation. 88 Genome-wide nucleosome positioning analysis in Arabidopsis thaliana and human cells also showed that DNA methylation is enriched in nucleosome-bound DNA rather than linker DNA, indicating that nucleosome-bound DNA is a preferred substrate for DNA methylation. 83 Nucleosome positioning displays a sequence preference characterized by particular dinucleotides, tending to occur periodically throughout the nucleosome with 10-bp periodicity. 90 Dinucleotide preference might arise from the near-circular wrapping of the nucleosomal DNA, which requires sharp bending every helical repeat (10 bp).90,91 DNMTs enter the major groove to access and methylate the cytosine on the outside of the nucleosome (minor groove). As nucleosome-bound DNA shows a 10-bp periodicity in its CG, CHG, and CHH methylation, 83 nucleosomes appear to dictate access to the DNA and therefore set the register of methylation for all DNA methyltransferases. DNA methylation with 10-bp periodicity occurs in different regions of the Arabidopsis and human genomes, including genes, promoters, pericentromeric regions, and euchromatic arm regions, suggesting that the relationship between nucleosome positioning and DNA methylation is general. Nucleosomal DNA is believed to be much less accessible than linker DNA to many proteins that act on naked DNA to carry out essential functions. In vitro, DNMT3A shows preferential DNA methylation activity towards naked DNA and the linker regions of nucleosomal DNA, whereas DNMT3B has weak activity towards the nucleosome core region. 92 However, in vivo, methylation in nucleosome-spanning DNA is much more enriched than in linker DNA, in agreement with the finding that DNMT3A and DNMT3B are strongly anchored to nucleosomes rather than linker DNA. This result supports the view that nucleosomes are preferentially targeted by DNMTs in vivo. 82
Recent genome-wide studies have revealed that nucleosomes are significantly more enriched in exons compared to introns, consistent with other recent findings of enhanced exonic methylation of gene bodies. 93 With the development of next-generation sequencing, DNA methylation patterns over the whole genome have been analyzed thoroughly at single base-pair resolution from plants to humans. While gene body methylation is conserved, its function in these regions is far from clear. In mammals, tissue- and cell type–specific DNA methylation exists in a small percentage of 5′ CGI promoters, whereas the majority of DNA methylation occurs in intragenic or intergenic regions. DNA methylation is commonly regarded as a silencing mechanism more difficult to reverse than covalent histone modifications. Experimentally imposed gene body methylation of an integrated transgene revealed that methylation impaired elongation of transcription in vivo. 94 However, the results of genome-wide sequencing studies show that DNA methylation of gene bodies correlates with increased rather than decreased transcription2,89,95 (Fig. 3). Zemach et al. 89 analyzed DNA methylation patterns in 17 eukaryotic genomes and showed that gene body methylation is conserved between plants and animals. Promoter methylation is inversely correlated with gene transcription, whereas gene body methylation displays a roughly parabolic curve with transcription, showing that the most methylated genes are at around the 70th transcription percentile. 89 In human B cells, 17.5% of the CGIs at the 5′ ends of genes are methylated, which is much lower than those CGIs in gene bodies (35.7% of which are methylated). 96 The majority of all methylated CGIs are associated with genes (68%), and only 32% of all methylated CGIs are intergenic. 96 A positive correlation between intragenic methylation and transcription was detected in human B cells. It has been known for some time that housekeeping genes rarely have internal CGIs, whereas 49% of tissue-specific genes have such islands, which are often methylated. 97 Thus, Jones suggested that transcription might facilitate de novo methylation. 97 Another possibility is that intragenic methylation represses transcription of antisense transcripts (perhaps representing transcriptional noise) that would otherwise downregulate expression of the sense transcript 21 (Fig. 3). Lister et al. 2 also observed a widespread positive correlation between the mean methylation of gene bodies and transcriptional activity in human IMR90 fibroblasts. However, no such relationship was discernible in H1 ESCs. 2 These results suggest that DNA methylation has alternative roles in somatic cells compared to stem cells. Therefore, the positive correlation between gene expression and gene body methylation could be reinterpreted as a depletion of methylation in genes repressed during differentiation. The view that gene body methylation positively correlates with increased transcription, however, is challenged by Maunakea et al., who regard it as more a regulator of alternative promoter usage. 51 They discovered that CGIs in gene bodies overlapped significantly with marks of transcriptional initiation and that unmethylated CGIs also overlapped with trimethylated H3K4, a histone modification enriched at promoters. Moreover, for intragenic CGIs, the degree of DNA methylation correlated inversely with trimethylation of H3K4. All these findings, consistent with the transcription tags based on capped analysis of gene expression (CAGE) experiments, 98 indicate that intragenic CGIs function as alternative promoters, 34% of which exhibit tissue-specific methylation. For example, the CGIs ECR22 and ECR32, which are embedded within the body of the SHANK3 gene, functioned as promoters for the 22t and 32t genes, respectively, and the methylation status of the ECR22 and ECR32 promoters inversely correlated with transcription of their corresponding genes. The work of Maunakea et al. strongly supports an earlier proposal that CpG islands are genomic footprints of promoters and that their methylation contributes to stable long-term silencing of the associated genes. 99 However, the finding that intragenic methylation contributes to gene silencing 51 is seemingly contradictory to others’ conclusions that gene body methylation correlates with increased transcription.2,89,95,96,100 The discrepancy may arise from the different methods of measuring DNA methylation and transcription since some groups use the average methylation level over the entire gene body rather than examining specific CpG sites with potential regulatory function. In addition, previous traditional or “canonical” gene expression measurements did not discriminate between which transcripts were being measured when multiple overlapping transcripts were present. 51 Taken together, intragenic CGIs might function as alternative promoters in a cell- or tissue-specific manner, although their methylation appears to positively correlate with the level of transcripts measured via traditional methods (Fig. 3). Nonetheless, a fundamental unanswered question remains as to whether all intragenic CGIs function as alternative promoters. If not, then the impact of gene body methylation on transcription in loci where no alternative promoters exist should be thoroughly investigated in the future and may tie in with regulation of transcriptional elongation as reported by Lorincz et al. 94
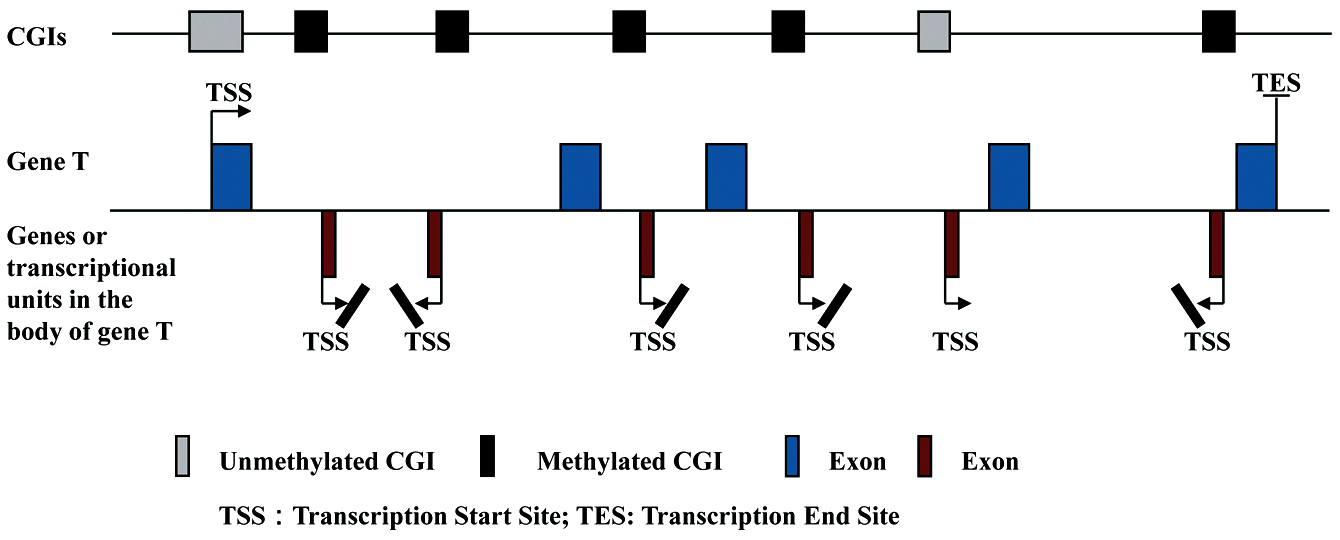
Intragenic CpG islands (CGIs) function as alternative promoters. CGIs embedded in the body of gene T may function as alternative promoters, whose methylation inversely correlates with the transcriptional levels of their corresponding genes. However, the total average level of intragenic DNA methylation appears to display a positive correlation with the transcription of gene T.
DNA Demethylation
In mammals, DNA demethylation also plays an important role in development and tumorigenesis. DNA demethylation, occurring in primordial germ cells (PGCs) and in early embryos, is essential for cells to return to a pluripotent state. The greatest loss of methylation in PGCs is observed within introns, intergenic regions and repeats, followed by exons, and then promoters. 101 Long terminal repeat (LTR)–ERV1 and LTR-ERVK elements, however, retain high levels of methylation. 101 In cancer, global genomic hypomethylation, leading to genomic instability and oncogene activation, affects repetitive sequences, imprinted genes, tissue-specific genes, oncogenes, and genes associated with invasion and metastases, 102 whereas many tumor suppressor genes are hypermethylated and silenced. 22 Active demethylation in plants is carried out by 5-methylcytosine (5mC) DNA glycosylases such as Demeter and Demeter-like proteins.103,104 However, this class of enzymes has not been identified in mammals. The DNA methylation erasure mechanisms in mammals are still poorly understood and controversial, although a number of potential mechanisms have been proposed.
Demethylation may be carried out by cytosine deaminases, converting 5mC to thymine, followed by T-G mismatch repair that specifically replaces thymine with cytosine. The recent discovery that activation-induced cytosine deaminase (AID) deficiency affects global DNA demethylation in murine PGCs 101 and that AID is required to demethylate pluripotency-associated genes during reprogramming of a somatic genome 105 demonstrates that AID has a substantial role in erasure of DNA methylation in vivo. AID and the related deaminase apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 (APOBEC1), both have robust 5mC deaminase activity in vitro, resulting in T-G mismatches in DNA that are effectively repaired through the base excision repair (BER) pathway.106,107 AID and MBD4 are involved cooperatively in demethylation of DNA 108 ; this link might also involve GADD45, which has also been implicated in demethylation of DNA. 108
In addition to assistance in recruitment of the DNA demethylation machinery to methylated DNA via its methyl- CpG–binding domain, MBD4 also possesses base excision repair DNA glycosylase activity, as well as 5-methylcytosine DNA glycosylase activity. 109 DNA demethylation in vivo may, in part, be mediated directly by MBD4 without the need for AID/APOBEC. Parathyroid hormone (PTH) induces active demethylation of the 5mC sites in the CYP27B1 promoter in human 293F cells. 110 Chromatin immunoprecipitation (ChIP) analysis showed that MBD4 was strongly bound to the CYP27B1 promoter, and knockdown of MBD4 blocked PTH-induced demethylation of 5mC at this promoter. 110 Activated MBD4 by PTH stimulation promotes incision at methylated DNA regions through its glycosylase activity, 111 and a base excision repair process appears to complete DNA demethylation at the MBD4-bound promoter. Such PTH-induced DNA demethylation, and the subsequent transcriptional derepression, is impaired by Mbd4 knockout. 110 The above findings therefore suggest that MBD4 is indispensable for PTH-induced DNA demethylation in the CYP27B1 promoter in human cells. However, the generalizability of these findings to the whole genome has not yet been investigated.
Most recently, it was demonstrated that oxidation of 5mC by TET family hydroxylases may also participate in active DNA demethylation.112,113 5-hydroxymethyl-2′-deoxycytidine (hmC) exists in murine brain tissue and cultured ESCs, which is formed via oxidation of the methyl group catalyzed by the hydroxylase TET1.112,113 It is unlikely that the hmC is a product of DNA damage since no DNA damage products, such as 8-oxoguanine, a preferential target for oxidants, or thymidine glycol, produced in vitro by the oxidation of 5mC, were detected in the same tissue or cells in vitro or in vivo. Conversion of 5mC to hmC may facilitate passive DNA demethylation by excluding the maintenance DNA methyltransferase DNMT1, which recognizes hmC poorly. 114 HmC may also be an intermediate in a pathway of active DNA demethylation, supported by the finding that a glycosylase activity specific for hmC exists in bovine thymus extracts. 115
Conclusion
Recent advances and significant ongoing research efforts aimed at characterizing the epigenome and its regulation place us firmly in an epigenetic era after the genetic era that culminated in sequencing of the human genome. The term “epigenetics,” which literally means “outside conventional genetics,” refers to the study of heritable changes in gene expression that occur independent of changes in primary DNA sequence. Epigenetic modifications are essential for mammalian development and cell proliferation, but they become disrupted in mature mammals, either by random factors or environmental influences. 116 Disruption of epigenetic processes results in altered transcriptional states that can culminate in malignant cellular transformation. Epigenetic states may be altered by environmental factors, likely leading to the development of abnormal or pathological phenotypes. A particular epigenetic state is maintained by epigenetic modifications, which include DNA methylation, posttranslational modifications of histone proteins, noncoding RNAs, as well as nucleosome positioning along the DNA. This raises two general questions: What are the roles of these epigenetic marks, and how are they coordinated in normal development and disrupted in disease? Being recognized as a key regulator of transcriptional stability, DNA methylation establishes a silent chromatin state by collaborating with other proteins that modify nucleosomes. Analysis of DNA methylation profiles will therefore enhance our understanding of the entire epigenome. The integration of DNA methylation with other epigenetic modifications is clearly a complex process that depends on the collaboration of numerous components, many of which remain to be elucidated.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the