
Abstract
The endoplasmic reticulum (ER) is an essential organelle involved in many cellular functions including protein folding and secretion, lipid biosynthesis, and calcium homeostasis. Proteins destined for the cell surface or for secretion are made in the ER, where they are folded and assembled into multi-subunit complexes. The ER plays a vital role in cellular protein quality control by extracting and degrading proteins that are not correctly folded or assembled into native complexes. This process, known as ER-associated degradation (ERAD), ensures that only properly folded and assembled proteins are transported to their final destinations. Besides its role in protein folding and transport in the secretory pathway, the ER regulates the biosynthesis of cholesterol and other membrane lipids. ERAD is an important means to ensure that levels of the responsible enzymes are appropriately maintained. The ER is also a major organelle for oxygen and nutrient sensing as cells adapt to their microenvironment. Stresses that disrupt ER function lead to accumulation of unfolded proteins in the ER, a condition known as ER stress. Cells adapt to ER stress by activating an integrated signal transduction pathway called the unfolded protein response (UPR). The UPR represents a survival response by the cells to restore ER homeostasis. If ER stress persists, cells activate mechanisms that result in cell death. Chronic ER stress is increasingly being recognized as a factor in many human diseases such as diabetes, neurodegenerative disorders, and cancer. In this review, we discuss the roles of the UPR and ERAD in cancer and suggest directions for future research.
The Unfolded Protein Response
Multiple perturbations can cause accumulation of unfolded proteins in the endoplasmic reticulum (ER) and activate the unfolded protein response (UPR). 1 These conditions include hypoxia, glucose deprivation, oxidative stress, viral infection, high fat or cholesterol, and mutations in specific proteins. The UPR regulates transcription and translation of genes in an attempt to re-establish homeostasis and restore ER functions. The UPR relieves ER stress by inducing genes such as ER chaperones to increase the protein-folding capacity of the ER, by up-regulating components of ERAD pathway to enhance the clearance of unfolded proteins from the ER, and by inhibiting general protein translation and specifically translation of proteins in the ER. While the UPR initially aims to promote cell survival, if ER stress persists or is prolonged, it also activates pathways leading to cell death.
In higher eukaryotes, the UPR is generally considered to involve at least 3 signaling pathways emanating from the ER. Among the ER stress sensors are IRE-1 (inositol-requiring protein 1), PERK (PKR-like ER kinase), and ATF6 (activating transcription factor 6); all 3 are integral ER membrane proteins. The activation of these proximal sensors is believed to be dependent on dissociation from the ER chaperone BiP, which usually prevents their dimerization and activation. 2,3 As unfolded proteins accumulate in the ER, BiP is sequestered from these sensors, allowing their oligomerization and activation. IRE1 and PERK are activated by homodimerization and phosphorylation. ATF6 is transported to the Golgi apparatus, where it undergoes regulated intramembrane proteolysis (RIP) to release its cytoplasmic domain, which translocates to the nucleus to activate gene transcription. 3-5
The UPR clearly has both survival and cell death effects (Fig. 1). The mechanisms that determine cell fate during ER stress are not well understood. For example, short-term exposure to ER stress initially increases AKT signaling, but prolonged ER stress suppresses AKT signaling. 6,7 The activation time course for the 3 UPR pathways differs in ER stress. 8 IRE1 activity is usually rapidly attenuated, whereas PERK and ATF6 activity is more prolonged. Sustained IRE1 activity promotes cell survival, suggesting that inactivation of IRE1 signaling sensitizes cells to the deleterious effects of chronic ER stress. 8 With this in mind, we will discuss the UPR sensors and their effects on cell fate determination.
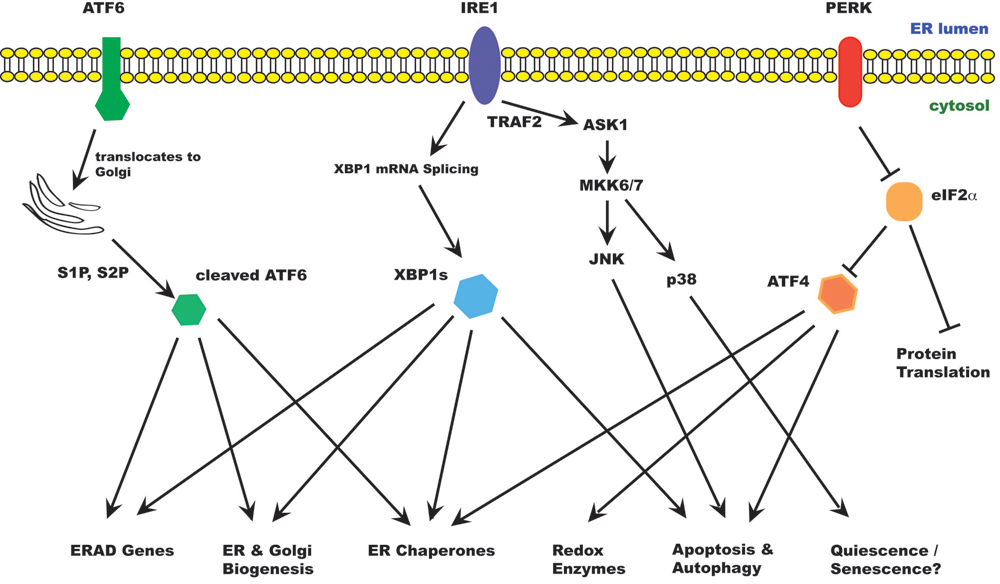
The unfolded protein response in mammalian cells. ER stress activates the unfolded protein response (UPR). The UPR acts to relieve ER stress, but prolonged UPR can also lead to cell death. There are at least 3 ER stress sensors on the ER membrane: IRE1α, PERK, and ATF6. Activated IRE1α splices an intron from XBP1 mRNA, producing XBP1s. XBP1 is a transcription factor that up-regulates many ER chaperones and genes involved in ERAD as well as membrane biogenesis. IRE1α also binds TRAF2 and activates ASK1 and downstream kinases, leading to activation of JNK and p38MAPK. JNK activation promotes autophagy and apoptosis; p38 promotes cell survival in a quiescence-like state. Activated PERK phosporylates eIF2α, resulting in inhibition of protein translation. However, some specific mRNAs, including ATF4, are translated under these conditions. ATF4 induces expression of genes involved in redox response, autophagy, and apoptosis. When activated, ATF6 translocates to the Golgi apparatus, where it is processed by Golgi proteases (S1P, S2P) to release the active transcription factor. ATF6 induces genes involved in ER homeostasis and membrane biogenesis.
IRE1 is a type I transmembrane protein of approximately 100 kDa and contains both a Ser/Thr kinase domain and an endoribonuclease domain. The substrates for IRE1 kinase activity are not known. Upon activation, IRE1 cleaves an intron of XBP1 (X-box–binding protein 1) mRNA. This splicing event produces a frame shift and results in the translation of the spliced form of XBP1, a 41-kDa basic leucine zipper (bZIP) family transcription factor that induces genes involved in UPR and ERAD. 9 IRE1 also cleaves many mRNAs that encode secreted proteins, reducing the load of protein in the ER, an activity that would favor restoration of ER homeostasis. 10,11
IRE1 signaling is activated early during ER stress and rapidly attenuated. Attenuation of IRE1 signaling occurs when its cytosolic domain interacts with that of BAX inhibitor-1 (BI-1), a 6–transmembrane domain containing protein in the ER. 12-15 Binding of BI-1 to IRE1 inhibits its endoribonuclease activity and modulates XBP1 splicing under conditions of mild ER stress. 15 Moreover, the unspliced form of XBP1 (XBP1u) can form a heterodimer with XBP1s and increases its degradation, thereby regulating the duration of its effects. 16 Finally, IRE1 is normally stabilized by HSP90 but rapidly degraded when cells are treated with geldanamycin. 17 The dynamics of HSP90/IRE1 interaction can therefore potentially determine the kinetics and amplitude of the IRE1 response and influence cell fate decisions during ER stress.
IRE1 binds the RING finger protein TRAF2 and activates ASK1 (apoptosis signal–regulating kinase 1), leading to activation of JNK and p38. The IRE1-ASK1-JNK pathway has been implicated in ER stress–induced cell death. 18,19 Expression of antiapoptotic BCL-2 family proteins such as BCL-2 and BCL-XL has been shown to inhibit JNK activation and reduce apoptosis in ER stress. Conversely, JNK has been reported to activate the proapoptotic BCL-2 family protein BIM and inhibit BCL-2. 20,21 JNK phosphorylation of BCL-2 activates Beclin-1, resulting in increased autophagy. 22 Autophagy is normally a survival response to nutrient deprivation but can also induce cell death independent of apoptosis. In murine cells, IRE1 activates Caspase 12, which activates a cascade of caspases leading to cell death. 23 It has been suggested that in human cells, CASP4 may play a similar role to Caspase 12, although its significance in the majority of cell types is not clear. 24
PERK is also a type I transmembrane protein kinase in the ER. Oligomerization of PERK induces its autophosphorylation and activates its Ser/Thr kinase activity. Activated PERK phosphorylates eIF2α (eukaryotic initiating factor 2 subunit α), thereby repressing protein translation. 25 However, certain mRNAs are translated preferentially under these conditions. One of these genes is ATF4, the translation of which is normally hindered by its uORF (upstream open reading frame). When eIF2α is inhibited, initiation at the start codon of ATF4 can occur, allowing for successful translation. 26 ATF4 promotes expression of ER chaperones, genes involved in glutathione synthesis and resistance to oxidative stress and genes involved in amino acid metabolism and transport. 27 On the other hand, ATF4 also induces CHOP (C/EBP homologous protein), which plays an important role in ER stress–induced cell death. CHOP can exacerbate ER stress by increasing the ER load and by inducing CHOP expression of the ER oxidase ERO1α, which makes the ER lumen more oxidative. CHOP has also been reported to induce expression of BIM and repress the expression of BCL-2, 28,29 further favoring cell death.
ATF6 is a type II transmembrane protein that is activated by RIP. During ER stress, ATF6 is transported to the Golgi apparatus, where it is cleaved sequentially by the Golgi resident serine proteases S1P and S2P (site 1 and site 2 proteases, respectively) to release its cytosolic domain. 30 The 50-kDa bZIP-containing cytosolic domain translocates to the nucleus to induce expression of CHOP, ER chaperones, and ERAD components, notably SEL1L, Herp, the ubiquitin ligase Synoviolin (described below), and EDEM1 (ER degradation enhancing α mannisidase-like protein 1). 31-33 ATF6α is required to facilitate recovery from acute stress and tolerance to chronic stress. ATF6α-null animals show increased propensity to organ dysfunction in vivo when challenged with chemical inducers of ER stress. 31 ATF6α is also capable of activating phosphatidylcholine synthesis to support ER biogenesis independently of XBP1. 34 ATF6α phosphorylation has been suggested to induce BiP expression under certain conditions, such as upon treatment with phosphatase inhibitor or exposure to the amino acid analog azetidine, which causes protein misfolding. 35 Under these conditions, ATF6α activation seems to be dependent on phosphorylation consequent to p38 signaling. Interestingly, ATF6α interferes with SREBP2 transcriptional activity, hence suppressing lipid biosynthesis during glucose deprivation. 36
UPR and Cancer
UPR and Tumor Cell Survival
In rapidly growing tumors, cancer cells face harsh microenvironments characterized by hypoxia, nutrient deprivation, acidosis, and nonpermissive interactions with stromal cells and extracellular matrix. These environmental stressors induce ER stress, and cells respond by activating the UPR.
The UPR plays an important role in cancer cell survival in hypoxia. The IRE1-XBP1 pathway has been shown to promote tumor growth in xenograft models. 37 Depletion of XBP1 results in cell sensitization to ER stress–induced cell death and smaller tumors when injected subcutaneously into immuno-compromised mice. Expression of XBP1s restores tumor growth under these conditions, suggesting that the IRE1-XBP1 axis is important for tumor cell survival and growth in hypoxic conditions. Loss of XBP1 also sensitizes cells to death from oxidative stress. 38 XBP1-deficient cells show more extensive ROS generation and prolonged p38 activation. Knockdown of XBP1 reduces catalase expression and enhances ROS generation, which can be rescued by extrinsic catalase. 38 This effect can be explained, at least in part, by the finding that XBP1 enhances the expression of catalase. 38
A role for the IRE1-XBP1 pathway is further supported by in vivo bioluminescence imaging of tumor cells expressing a XBP1-luciferase reporter in which luciferase is expressed only when XBP1 is spliced by activated IRE1. 39 XBP1 splicing can be detected even in relatively small tumors, suggesting that ER stress occurs throughout tumor growth. 39 Similar imaging studies in transgenic mice show that XBP1 splicing occurs during primary tumor growth in several genetic models for breast cancer. 39 The levels of XBP1 activity differ between tumors, correlating with their growth rate and inversely with glucose availability, suggesting IRE1 activation in response to glucose starvation. 39 These studies raise the question of whether IRE1 activation is responsible for faster tumor growth or merely a reflection of faster growing tumors.
Another important sensor in UPR is PERK, which phosphorylates eIF2α upon activation. PERK activation leads to cell cycle arrest in G1. 40 The induction of cell cycle arrest and inhibition of protein translation allow cells to conserve energy and survive during stressful conditions such as hypoxia and ischemia. PERK is essential for tumor cell development of hypoxia tolerance. Compared to PERK+/+ tumors, PERK–/– tumor cells show reduced viability under hypoxic conditions and form smaller tumors and are defective in their capacity to stimulate formation of functional blood vessels. 41
PERK activation increases the translational efficiency of ATF4, which induces multiple stress response genes including genes involved in resistance to oxidative stress. PERK also phosphorylates Nrf2, promoting its stability by dissociation from Keap1. 42,43 Nrf2 is a bZIP transcription factor that induces expression of the ARE (antioxidant response elements) genes including antioxidants and detoxification enzymes, immune signaling, protein trafficking, protein degradation, cell growth and survival, and the chaperone system. 44,45 Nrf2 also inhibits expression of CHOP, providing one way of reducing cell death induced by PERK activation. 42,46 Like Nrf2, ATF4 also induces ARE-containing genes. 27 Thus, PERK activation converges on the up-regulation of ARE-containing genes through both Nrf2 and ATF4 and thereby enhances cellular defense against oxidative stress. Knockdown of PERK in human breast cancer cells results in cell cycle arrest in G2/M. 47 The G2/M arrest is likely due to reduced Nrf2 activity in PERK-deficient cells, resulting in accumulation of ROS and oxidative DNA damage and subsequent activation of DNA double strand–break checkpoint. 47 Loss of PERK delays tumorigenesis in the MMTV-Neu model for breast cancer and seems to reduce the incidence of pulmonary metastases in these models. 47 On the other hand, conditional deletion of PERK in mouse mammary tissue results in a higher incidence of spontaneous mammary adenocarcinoma. 47 Thus, PERK deletion activates DNA damage response activation and limits tumor growth, but long-term accumulation of DNA damage makes PERK–/– mice more susceptible to spontaneous tumorigenesis.
Transient exposure to ER stress can condition cells for survival during a subsequent, more severe stress. This preconditioning is likely due to induction of prosurvival genes that prepare the cells for the subsequent insult. As tumor cells in the primary tumor are exposed to hypoxia, they might be preconditioned to survive the subsequent metastatic process. This is consistent with the more aggressive phenotype of more hypoxic tumors. Inhibiting the UPR may not only inhibit survival of cancer cells within the primary tumor but could also potentially reduce their ability to survive the subsequent stresses during metastasis.
UPR and Tumor Dormancy
The UPR is also implicated in tumor cell survival during dormancy. Tumor dormancy is a protracted stage in tumor progression during which tumors remain asymptomatic for an extended period of time. Dormant tumors are difficult to detect but can be activated to become rapidly growing tumors when conditions are favorable, presenting a problem for clinical treatment of the residual disease. Tumor dormancy is usually related to insufficient angiogenesis and a balance of proliferation and apoptosis within the tumor. 48-50 In some cases, tumor dormancy is caused by tumor cell quiescence.
Studies in animal models have shown that many tumor cells remain in distant organs as single dormant cells. 51 This is regulated in part by tumor cell interactions with the extracellular matrix and a balance of p38 over ERK activation. 52,53 In a study demonstrating the role of p38 in squamous carcinoma dormancy, high levels of uPAR (urokinase receptor for plasminogen activator) on the cell surface are correlated with growth of tumor cells, whereas p38 is implicated in cell arrest in a G0-like quiescent state. In this state, the quiescent cells are resistant to DNA damaging agents. 52,53 A subsequent study comparing a squamous carcinoma T-HEp-3 and its dormant derivative D-HEp3 suggests that high PERK-eIF2α signaling is responsible for survival of D-HEp3 cells but also induces their arrest in G0/G1. 54 Expression of PERK in T-HEp3 arrests these cells in G0/G1 and inhibits tumorigenesis in subcutaneous xenograft models and chicken embryo chorioallantoic membrane (CAM) system. The chicken embryo CAM is highly vascularized so early tumor growth is not completely dependent on neovascularization in this model, indicating that cellular dormancy is responsible for inhibition of tumor growth. 54 PERK has been shown to arrest cells in G1 by its repression of protein translation and the resulting reduction in cyclin D1 levels. 40 Moreover, ATF6 activation is essential for long-term survival of dormant cells and resistance to chemotherapy, nutrient deprivation, and microenvironmental stresses in D-HEp3 cells. 55 In this model, ATF6 activation depends on MKK6 and p38 signaling. ATF6 increases the expression of Rheb, which activates mTOR activity independently of AKT. 55 ATF6 signaling therefore enables tumor cell survival during prolonged periods of dormancy and may be a viable target for eradicating dormant tumor cells. MKK4, a MAP3K activating both JNK and p38, has been shown to suppress metastasis of ovarian cancer cells by activating p38 signaling and inducing cellular senescence. 56 p38 signaling has been associated with cellular senescence during oncogenic transformation. 57 Indeed, slight changes in p38 activity can suppress tumorigenesis. 58 An interesting hypothesis is that UPR activation allows cells to escape p38-induced senescence and relief from UPR allows cells to switch to a rapidly proliferating state.
Distinct from cellular dormancy, tumor dormancy can also result from failure of the tumor to grow as a result of apoptosis or inability to initiate vascularization. The ability to recruit and build new vasculature is critical for tumor growth. Once the tumor grows to a certain size, availability of oxygen and nutrients becomes limited by diffusion, resulting in local hypoxia and ischemia. Cells adapt to the hypoxic conditions, up-regulating HIF (hypoxia inducible factor), which reprograms the cell metabolism and promotes cell survival (reviewed in Semenza 59 ). Hypoxia is also a potent inducer of the UPR. These transcriptional and translational adaptations are important for restricting global protein synthesis while promoting translation of genes important for angiogenesis and cell survival. 41,60 The UPR, through IRE1, PERK, and ATF6, also induces transcription of VEGF, which allows for survival of rapidly growing tumor cells. 61-63 Targeting the UPR at this stage could possibly impair translation of HIF and VEGF, thereby suppressing cancer cell survival and limiting neoangiogenesis to support tumor growth.
UPR and Differentiation
The UPR is induced during neuronal differentiation of stem cells including mouse embryonic stem cells. 64 Conversely, treatment of stem cells with a chemical inducer of ER stress induces differentiation and the expression of neuron-specific genes. 64 The UPR has also been associated with cellular dedifferentiation. ER stress may induce dedifferentiation in those cells whose phenotype is associated with the synthesis of a large amount of proteins in the ER. In thyroid cells, thapsigargin and tunicamycin, both of which induce ER stress and the UPR, induce dedifferentiation. Dedifferentiation of these cells is also associated with changes in gene expression consistent with an epithelial-to-mesenchymal transition, an important early step in metastasis. 65 In breast cancer cells, overexpression of CK19 (cytokeratin 19) causes cell cycle arrest, reduced cell motility, and increased drug resistance. 66 CK19 expression activates p38/ PERK and down-regulates ERp29 (ER protein 29), which induces a mesenchymal-to-epithelial transition when overexpressed in breast cancer cells. 66
UPR and Cell Death
Apoptosis of cancer cells at the secondary site is a major limiting step in the metastatic cascade. The exact mechanism for apoptosis is not well characterized, although it is likely to depend on the cell types and the organs studied. The IRE1-ASK1-JNK axis is associated with apoptotic cell death resulting from ER stress. 18,19 Interestingly, MKK4, a MAP3K upstream of JNK, has been found to suppress metastasis in prostate cancer by activation of JNK. 67,68 Transfection of MKK4 in prostate cancer cells that lack MKK4 expression suppresses metastasis but not growth of the primary tumor. 68 Differential activation of JNK during metastasis presumably underlies the differences on primary tumor growth compared to metastasis, although the nature of the stimulus is not clear. 67 The role of JNK in metastasis-associated cell death has not been well studied. JNK is required for cell transformation. However, persistent activation of JNK can lead to cell death by apoptosis as well as autophagy. 22
Loss of adhesion from normal substratum, which occurs as cancer cells enter the circulation during metastasis, usually initiates apoptosis (anoikis). Loss of adhesion has been shown to activate PERK, although it is not clear whether other arms of the UPR are activated. 69 Moreover, impaired mitochondrial function increases ROS generation and activates the UPR. 70 As cells begin metastasis, the redox microenvironment changes drastically. The relations between the redox environment and differential JNK activation by MKK4 during metastasis will be an interesting area for investigation.
Transient acute exposure to ER stress can condition and prepare cells for survival during a subsequent, more severe stress. This “preconditioning” is likely due to induction of prosurvival genes that prepare the cells for the subsequent insult. Analogously, tumor cells in the primary tumor are exposed to hypoxia and might be preconditioned to survive the subsequent metastatic process. Inhibiting the UPR may not only inhibit survival of cancer cells within the primary tumor but could also potentially reduce their ability to survive the subsequent stresses during metastasis.
Ubiquitylation and ER-Associated Degradation
One important consequence of the UPR is the up-regulation of ERAD components. ERAD plays an important role in ER homeostasis by eliminating misfolded proteins, protein subunits that fail to assemble into their native complexes, and proteins whose levels must be acutely regulated in response to metabolic needs. The UPR and ERAD are functionally coupled to ensure optimal cell function and survival. 71 Genes integral to ERAD are up-regulated by ER stress and the activation of the UPR. For this reason, an intact UPR is necessary for efficient ERAD. This dynamic relationship is exemplified in B cells where ER stress consequent to the amount of protein entering the secretory pathway leads to ER stress, UPR, and the increased synthesis of ER membrane and ERAD components. 72
One can conceptualize ERAD as 3 interdependent components: 1) recognition of the protein target and its association with the appropriate luminal chaperones, 2) association with the ERAD ubiquitylation machinery, and 3) retrotranslocation into the cytosol for degradation by proteasomes. The proteins responsible for each of these 3 aspects are subjects of intense research. Herein, we focus primarily on the ubiquitylation machinery involved in ERAD and their roles in cancer and touch only briefly on other aspects of this process, which are reviewed in detail elsewhere. 73,74
The signals for substrate recognition by the ERAD machineries are complex. With the exception of regulated proteins, ERAD substrates are generally considered misfolded or otherwise unassembled into native multiprotein complexes. It is not clear how these substrates are distinguished from those in the normal process of protein folding. A simplistic view is that substrates are associated with molecular chaperones through both exposed hydrophobic patches and N-linked oligosaccharides while attempts at correct protein folding occur mediated by chaperones. The chance of being targeted may therefore be determined stochastically based on the duration of chaperone association. For ER proteins that form multi-subunit complexes, their assembly likely masks hydrophobic patches or other sequences recognized by the ERAD machineries.
ERAD in Saccharomyces cerevisiae
Studies in S. cerevisiae suggest that the location of misfolding within the substrate dictates the pathway for its degradation. There are 2 ubiquitin ligase (E3) complexes responsible for ERAD in yeast known as the HRD1/DER3 and DOA10 complexes (reviewed in Kostova et al. 75 ).
The HRD1/DER3 complex is centered around Hrd1p/Der3p. This protein was discovered independently in genetic screens for proteins that inhibit degradation of Hmg2 (Hrd1p) and in a screen for inhibition degradation from the ER in general (Der3p). The Hrd1p/Der3p ubiquitin ligase is a 6–transmembrane RING finger protein that interacts with Ubc1p, Ubc6p, and Ubc7p to catalyze the polyubiquitylation of substrates. 76 RING (really interesting gene) fingers are 40-to-60–residue, Cys-rich motifs that bind 2 zinc ions and that for the most part have ubiquitin ligase activity. 77 Ubc1p is a soluble E2 (ubiquitin-conjugating enzyme) with a C-terminal ubiquitin-association domain (UBA). The UBA adopts a 3 α helix that binds ubiquitin. 78 The mammalian ortholog, E2-25K, is notable for its capacity to form proteasome-targeting K48-linked polyubiquitin chains independently of E3 (ubiquitin ligase). 79 Ubc6p, which contains a C-terminal ER anchor, is the only transmembrane E2 in yeast. 76
Ubc7p is also cytosolic but is specifically recruited to the ER membrane by an accessory protein common to both of the yeast ERAD complexes known as Cue1p. 80 This 203 aa protein, which is inserted into the ER through its N-terminal transmembrane domain, binds Ubc7p. 80 The binding site has recently been identified as a 53-residue region (aa 151-203) known as the Ubc7-binding region (U7BR). 81 Cue1p initially appeared to be simply a means of recruiting Ubc7p to the ER membrane. However, recent studies have demonstrated that the U7BR can enhance Hrd1p-mediated ubiquitylation in vitro and interestingly, and of unknown significance, is the finding that a more extended region of Cue1p can activate E2 to mediate RING finger–independent ubiquitylation, at least in vitro. 81,82 Cue1p is also notable for having a CUE domain, which is a ubiquitin-binding domain structurally analogous to the more prevalent UBA domain. The CUE domain of Cue1p binds ubiquitin weakly. To date, the CUE domain appears dispensable for Cue1p function in ERAD or ER stress response.
The HRD1/DER3 complex also includes other factors necessary for ERAD. Hrd3p, a single-pass transmembrane ER protein, is required to stabilize Hrd1p/Der3p. 83 The Hrd3p luminal domain associates with an ER lectin Yos9p, which together with the yeast Kar2p (homolog of mammalian BIP) recruits the substrate for presentation to Hrd1p/Der3p. 84-87 Another factor necessary for ERAD is Usa1p. 88 Usa1p spans the ER membrane twice, with both its N- and C-termini facing the cytosol. The N-terminus of Usa1p has a UBL (ubiquitin-like) domain, which is required for its function. Herp, the mammalian homolog of Usa1p, is strongly induced by UPR and required for ERAD during ER stress. Usa1p likely serves to recruit Der1p, another component of the Hrd1p/Der3p complex. 89 The roles of Der1p are not clear, but its mammalian homolog, Derlin-1, has been suggested to function as part of the retrotranslocation complex. 90,91
The DOA10 ubiquitin ligase complex, which is centered on the multimembrane-spanning RING finger protein Doa10p, utilizes Ubc6p and Ubc7p for polyubiquitylation of ERAD substrates. 92 Interestingly, this E3 also utilizes the E2 pair Ubc4p/Ubc5p for ubiquitylation of other substrates, most notably the yeast mating factor MATα2. An interesting aspect of Doa10p is the finding that it is distributed both in the ER membrane and on the nuclear envelope, suggesting roles in multiple cellular compartments. 93
A central issue in ERAD, as noted above, is how substrates are targeted to specific ubiquitin ligases. Consistent with its association with Hrd3p and Yos9p, evidence suggests that Hrd1p/Der3p responds to “lesions” in the ER lumen or transmembrane segments, degrading substrates via the ERAD-L (ERAD-luminal) and ERAD-M (ERAD-membrane) pathways. 94 In contrast, Doa10p recognizes degradation determinants on the cytosolic side of the ER membrane or in the nucleus and defines the ERAD-C (ERAD-cytosolic) pathway. Accordingly, Hrd1p/Der3p substrates are generally confined to the ER membrane or lumen, whereas Doa10p substrates are more broadly distributed. As more ERAD substrates are defined, it seems likely that rules regarding these divisions will need further refinement.
As mentioned above, the mammalian ortholog of Der1p has been suggested to function as part of a retrotranslocation channel. Such channels are postulated to help overcome thermodynamic barriers to movement of ERAD substrates through the ER membrane. Another candidate for the retrotranslocation channel is Sec61p, which is also a critical component of the “translocon” that cotranslationally imports proteins into the ER. 95 It remains to be determined whether either of these is the bona fide retrotranslocon, whether translocons might be heterogeneous and even include the transmembrane domains of ubiquitin ligases, or whether such channels even exist and are truly required to overcome thermodynamic barriers. In this regard, it is important to note that retrotranslocation of many proteins is tightly tied to a hexameric AAA ATPase known as Cdc48p, which exists in complex with cofactors Npl4p and Ufd1p, which are ubiquitin-binding proteins. 96-98 The Cdc48p complex is itself recruited to the ER membrane through its association with Ubx2p/Sel1p. 99-101 Ubx2p/Sel1p contains an N-terminal UBA, which is important for ERAD, and a C-terminal ubiquitin-like UBX (ubiquitin-regulatory X) domain, which acts as a Cdc48p-binding module. 101 Cdc48p may deliver substrates directly to proteasomes, although evidence suggests that in some cases the accessory factors, Rad23p and Dsk2p, which bind substrates through UBAs and associate with proteasomes through their UBLs, serve as shuttles between Cdc48p and the proteasome. 102 It is not clear that all ERAD substrates require Cdc48p; in these cases, the ubiquitylated substrate may be directly recognized by the proteasome. As the 19S cap of the proteasome also contains a ring of AAA ATPase, it may be that this serves as an alternative to Cdc48p in “racheting” proteins out of the ER membrane.
Less is known of associated proteins involved in Doa10p function, although Cdc48p is also implicated for some Doa10p substrates. 94 Cdc48p is not required for degradation of soluble substrates of Doa10p. 103
ERAD in Mammalian Cells
As expected, the ERAD machineries in mammals are more complex. The human genome encodes approximately 50 RING finger proteins with putative transmembrane segments, most of which we expect to localize to the ER. Moreover, several cytosolic ubiquitin ligases have been shown to function in mammalian ERAD (see below). The complexity and redundancy of mammalian ERAD may cause the distinct ERAD pathways to overlap so that the rules regarding pairing of substrates in yeast are blurred in mammalian cells. In addition, at least 2 deubiquitylating enzymes (DUBs) have been shown to be involved in mammalian ERAD. Ataxin-3, the protein mutated in Machado-Joseph disease, is a DUB that can trim polyubiquitin chains from ERAD substrates, thereby modulating their degradation. 104 Another DUB, Usp19, is induced by ER stress and localizes to the ER. 105 Usp19 expression increases the levels of TCRα and mutant CFTR (cystic fibrosis transconductance regulator) consistent with its role in ERAD regulation. Mammalian ERAD does share some common features with that in yeast. The 2 homologs of Ubc6p, Ube2j1/NCUBE-1 and Ube2j2/NCUBE-2, are anchored to the ER membranes by their C-terminal hydrophobic stretch. Both of these E2s have been implicated in ERAD. There are also 2 homologs of Ubc7p, Ube2g1 and Ube2g2, both of which are cytosolic proteins. Interestingly, despite more than 80% identity, Ube2g2 is extensively implicated in ERAD, whereas there is considerably less evidence for a role for Ube2g1. Like Ubc7p, Ube2g2 is recruited to the ER membrane by its association with the ER-resident ubiquitin ligase RNF45/gp78. 106,107 RNF45 is a multimembrane-spanning RING finger ubiquitin ligase related to Hrd1p/Der3p. Synoviolin, the other homolog of yeast Hrd1p/Der3p, is also involved in ERAD. Synoviolin associates with OS9 (mammalian ortholog of Yos9p), Herp, and Derlin-1 for its function. Recent experiments also identify Aup1 as an essential component of Synoviolin pathway. 108 Aup1 has substantial homology to yeast Cue1p, although its function in mammalian ERAD is not well understood. Efficient extraction of certain polyubiquitylated substrates in mammalian cells also requires p97/VCP, the mammalian homolog of Cdc48p. p97 can be recruited directly to ubiquitin ligases such as RNF45 or via UBX domain–containing protein. 109,110
Inhibition of ERAD induces UPR while preventing UPR from efficiently restoring cellular homeostasis, shifting the balance towards proapoptotic aspects of the UPR. Thus, targeting the ERAD pathway represents an alternative strategy to inhibit the prosurvival function of UPR. In the following section, we review mammalian ERAD pathways defined by specific ubiquitin ligases and discuss their potential roles in cancers.
ERAD and Cancer
RNF45/gp78/AMFR
The first ubiquitin ligase integral to the ER membrane shown to function in ERAD in mammalian cells is RNF45/gp78/AMFR. 107 Knockdown of gp78 by siRNA abolishes ERAD of several mammalian ERAD substrates in cells. These include the extensively studied test substrates T-cell receptor subunits (CD3-δ and TCR-α) and the Z variant of α1-antitrypsin. 106,111 Depletion of gp78 results in the accumulation of CD3-δ in the ER membrane, suggesting the gp78-mediated ubiquitylation is an early event preceding retrotranslocation of substrates into the cytosol. 106 gp78 is also implicated in the degradation of Apoliprotein B-100, Insig-1, and HMG-CoA reductase. 112-114 The degradation of both Insig-1 and HMG-CoA reductase has been reported to be sterol regulated, implicating gp78 as an important regulator of cholesterol metabolism.
RNF45/gp78 is an integral membrane protein with multiple transmembrane spans. gp78 and the human homolog of Hrd1p/Der3p (Synoviolin) share over 50% homology in their transmembrane regions. Computer algorithms predict 5 or 6 transmembrane spans in gp78. In addition to a RING finger, the C-terminus of gp78 also contains an extended area of homology to the cytoplasmic domain of yeast Cue1p and a binding site that specifically recruits Ube2g2, the mammalian homolog of Ubc7p. This specific Ube2g2-binding region consists of a 27–amino acid peptide (residue 574-600) referred to as G2BR (Ube2g2-binding region). Indeed, overexpression of G2BR alone in cells is sufficient to inhibit ERAD due to sequestration of Ube2g2. The Ube2g2-binding site is required for gp78 ubiquitin ligase activity in cells. Structural studies show that G2BR binds with high affinity (Kd ~21 nM) to Ube2g2 on the “backside” of Ube2g2, on a surface opposite from its active site. 115 This interaction region is also distinct from the sites of interaction with RING finger E3s and with ubiquitin-activating enzyme. The high binding affinity is a result of multiple contacts involving both salt bridges and hydrophobic interactions. 115 Intriguingly, G2BR induces allosteric changes in Ube2g2, increasing the affinity of Ube2g2 for the RING finger domain of gp78 almost 50-fold. 115 This change in affinity translates into more efficient transfer of ubiquitin from the active site of Ube2g2 to substrates in the presence of G2BR. 115 This enhancement is not specific to the gp78 RING finger as G2BR provided in trans enhances the ubiquitin ligase activity of several other ERAD ubiquitin ligases specifically with Ube2g2. As discussed above, the Cue1p protein of yeast also contains a Ubc7p-binding region (U7BR) that enhances ubiquitin transfer of Ubc7p. 81 Cue1p recruits Ubc7p to the ER and enhances Hrd1p ubiquitin ligase activity, suggesting a common mechanism of function for both E2 binding sites. The G2BR has also been reported as required for gp78-dependent assembly of a polyubiquitin chain on the active site of Ube2g2. 116,117 Polyubiquitin chain formation on Ube2g2 appears to be required for efficient polyubiquitylation of the model substrate Herpc (Herp, cytosolic domain) in vitro. 117 A region between the RING and CUE domain (amino acid 379-395) has been proposed to effect dimerization of gp78 essential for this reaction, although it is dispensable for gp78 ubiquitin ligase activity. 117 In this model, the G2BR provides a docking site for Ube2g2, while the polyubiquitin chain is being assembled by a gp78 dimer. Further studies are needed to understand the physiological significance of gp78 polyubiquitylation on the Ube2g2 active site.
Unlike Cue1p, which functions as part of the HRD1 and DOA10 ligase complexes, gp78 requires its ubiquitin-binding CUE domain for function in vivo. 106 One possible role for the gp78 CUE domain could be to recruit substrates ubiquitylated by other E3s, allowing gp78 to function as an “E4” in the ubiquitylation cascade. 118 Indeed, gp78 has been shown to function as an E4 together with RNF5/RMA1 in the ubiquitylation of mutant CFTR. 119 However, in the case of the T-cell receptor subunit CD3-δ, substrate recognition by gp78 does not depend on the CUE domain, although ubiquitylation of CD3-δ in cells requires this domain. Similarly, gp78 recognizes a SOD1 mutant (G94A) associated with amyotrophic lateral sclerosis via a region between the RING finger and CUE domain, whereas it binds ataxin-3 via its transmembrane regions. 120 The exact determinant for substrate recognition is likely context dependent, allowing gp78 to recognize a broad range of substrates.
While the CUE domain and Ube2g2-binding site are required for gp78 activity in vivo, the C-terminal p97-binding region appears to be largely dispensable for ERAD. 106,110,121 However, there are a number of other ways in which p97 can be recruited to the ERAD machinery. p97 can be directly recruited by polyubiquitylated substrates. Alternatively, proteins associated with RNF45 could recruit this chaperone for gp78-mediated ERAD. In this regard, Ubxd8 recruits p97 for extraction of Insig-1 from the ER membrane. 109
RNF45/gp78 has been previously considered a receptor for the autocrine motility factor (AMF; also known as glucose-6-phosphate isomerase). The AMF receptor (AMFR) was isolated by phage expression screening using a monoclonal antibody (3F3A) that stimulates tumor cell motility in vitro. 122 This 323–amino acid protein, now referred to as AMFR isoform 1, was predicted to have a single transmembrane segment. 122 The discrepancy between the predicted size of the recombinant protein (34-35 kDa) and the migration of the cellular species recognized by 3F3A (78-80 kDa) led to the postulation that this protein was extensively glycosylated, hence the name gp78. However, the single putative N-linked glycosylation site (N599) resides in the cytosolic domain within the G2BR region involved in Ube2g2 recruitment. AMFR isoform 2, encoding a 643–amino acid protein with a predicted molecular weight of 73 kDa, 123 is the protein referred to herein as RNF45/gp78 and commonly referred to as AMFR. AMFR was postulated to function as a G protein–coupled receptor (GPCR). 123 Comparison of the 2 mRNA sequences shows that isoform 1 is 5′ truncated with multiple deletions causing frame shifts that result in translation of mostly the 3′UTR of RNF45. It is not clear that the reported 78-kDa cell surface protein AMFR recognized by 3F3A is indeed RNF45.
More recently, we have shown that knockdown of gp78 inhibits sarcoma metastasis without affecting growth of orthotopic primary tumors. 121 Curiously, the prometastastic function of gp78 requires an intact ubiquitin ligase activity but not its putative AMFR or GPCR function as the prometastatic function can be reconstituted in gp78-deficient cells by expression of the fusion of a single ER-targeted transmembrane domain and the gp78 cytoplasmic tail. 121 Similarly, the p97 binding activity is not required for RNF45 function in metastasis. 121 This prometastatic activity is due in part to gp78 targeting the metastasis suppressor KAI1/CD82 for degradation. 121 KAI1 is a tetraspanin and belongs to the family of metastasis suppressor genes that suppress tumor metastasis without inhibiting growth of the primary tumors. 124 KAI1 is believed to suppress metastasis by multiple mechanisms (reviewed in Miranti 125 ) including induction of tumor cell senescence upon interaction with the endothelial cell surface protein DARC and activation of apoptosis through oxidative stress. 126,127 Consistent with gp78’s role in ERAD, cells with reduced levels of gp78 show increased sensitivity to ER stress–induced cell death. These cells also show increased apoptosis when they first reach the lungs following intravenous dissemination. Suppression of KAI1 partially restores survival of gp78-deficient cells. 121 The in vivo relevance of gp78 to KAI1 pathology is further confirmed by an inverse correlation between gp78 and KAI1 levels in human sarcoma samples. 121 Thus, gp78 promotes metastasis by enhancing tumor cell survival and by degrading the metastasis suppressor KAI1. The identification of RNF45 as a ubiquitin ligase for KAI1 raises the possibility that ubiquitin-mediated regulation of other metastasis suppressors can also have dramatic impact on cancer metastasis. Understanding how metastasis suppressors are regulated posttranslationally should open up new approaches to the treatment of metastatic disease.
Recently, it is reported that MMTV-gp78 transgenic mice develop mammary hyperplasia associated with reduced levels of KAI1. 128 The role of gp78 in tumorigenesis is not well studied. Overexpression of gp78 can induce transformation of NIH 3T3 fibroblast. 129 It is plausible that higher levels of gp78 promote cell survival and facilitate transformation. If this were true, cell transformation by gp78 likely requires its ubiquitin ligase activity.
As noted above, 2 substrates of gp78, Insig-1 and HMG-CoA reductase, are important proteins involved in lipid biosynthesis. Lipid biosynthesis is essential for rapid proliferation of cancer cells. How alterations in lipid biosynthesis mediate gp78 effects on tumorigenesis and metastasis has not yet been explored. Recent studies have also implicated gp78 in the regulation of CYP3A4, the major enzyme responsible for drug metabolism in the liver. 130 If these in vitro results held up in vivo, gp78 levels might be a source of variations in drug interactions and bioavailability in cancer therapy. Interestingly, analysis of the tumor stroma suggests gp78 levels as a potential factor in the genetic causes of cancer health disparity between breast cancer patients of African and European descent. 131 Differences in the tumor microenvironment are proposed to account for the health disparity observed. The role of gp78 and ERAD in regulating the tumor microenvironment deserves further investigation.
Synoviolin
Another ER-resident ubiquitin ligase implicated in ERAD is Synoviolin, which is a homolog of S. cerevisiae Hrd1p/Der3p. Synoviolin has been described as an ERAD ubiquitin ligase for TCR-α and CD3-δ as well as for the Parkin-associated endothelin-like receptor (Pael-R), a misfolded G protein–coupled receptor implicated in PARK2-related Parkinson disease. 132-134 Cytosolic SGK1 (serum- and glucocorticoid-induced kinase 1), which regulates cell survival under stress conditions, is targeted to the ER via an N-terminal hydrophobic sequence, leading to its rapid degradation by an ERAD pathway also involving Synoviolin. 135 Synoviolin has also been reported to ubiquitylate cytosolic p53. 136 Synoviolin functions with both UbcH5B and C (Ube2d2 and Ube2d3) as well as the ERAD E2 Ube2g2 in vitro, although its cognate E2 in cells has not been identified. 132-134,136 In contrast to Hrd1p/Der3p, there is no evidence that Synoviolin is involved in the rapid sterol-regulated degradation of HMG-CoA reductase in mammalian cells. It has, however, been shown to target HMG-CoA reductase for the relatively slow sterol-independent degradation. 132,133
Synoviolin has been implicated in the pathogenesis of rheumatoid arthritis. 137 Overexpression of Synoviolin in mice causes arthropathy with synovial hyperplasia. 137 Homozygous deletion of Synoviolin results in embryonic lethality, whereas heterozygous knockdown increases apoptosis of synovial cells and confers resistance to collagen-induced arthritis in mice. 137,138 Consistent with these observations, proliferating synovial cells in rheumatoid arthritis overexpress Synoviolin and acquire resistance to ER stress–induced apoptosis in vitro. 137 Supporting its role as an antiapoptotic protein through ERAD, Synoviolin reduction by siRNA sensitizes rheumatoid synovial cells to ER stress–induced cell death and inhibits their ex vivo proliferation. 139
The role of Synoviolin in cancer is not clear. Synoviolin is capable of promoting the degradation of gp78 independently of gp78 ubiquitin ligase activity. 140,141 Consistent with this observation, Synoviolin-null cells have higher steady-state levels of gp78. 141 Furthermore, increasing Synoviolin levels results in accumulation of RNF45 substrates. 140,141 The physiological functions of this regulation are not well understood, but it is conceivable that Synoviolin regulates the levels of gp78 and hence the levels of its substrates in response to cellular metabolic needs. For example, the ubiquitin ligase activity of Synoviolin and gp78 towards their substrates is increased, while their self-ubiquitylation is reduced during UPR. 142 Unlike RNF45/gp78, Synoviolin is induced during UPR. 142,143 Increased levels of Synoviolin can function to reduce the levels of gp78 in this situation. It would be interesting to determine whether Synoviolin functions as a metastasis suppressor by down-regulating the levels of gp78. Synoviolin-null cells could be useful tools for dissecting the relative contributions of ER stress and KAI1 towards gp78-mediated metastasis and more broadly address the roles of ER stress in metastasis.
As mentioned above, Synoviolin targets Pael-R for degradation. 134 Pael-R is an ER protein found to be degraded by Parkin, a gene linked to genetic cases of Parkinson’s disease and a putative tumor suppressor. 144 This finding suggests that Synoviolin and Parkin may function in the same pathway. 134 Whether Synoviolin plays a role in Parkin tumor suppressor function awaits determination.
TRC8/RNF139
TRC8 was originally identified as a tumor suppressor that was lost as a result of chromosomal translocation t(3;8)(p14.2;q24.1) associated with hereditary renal cell carcinoma. 145 TRC8 is a 664–amino acid protein localized to the ER and contains multiple membrane-spanning domains including a sterol-sensing domain. 146 Overexpression of TRC8 in kidney cells suppresses growth of kidney cells in vitro and tumor formation in xenograft models in a RING-dependent manner due to a G2/M arrest and increased apoptosis. 147 TRC8 promotes its own ubiquitylation and degradation in the presence of sterols. 148,149 In the absence of sterols, TRC8 is stable and accumulates in cells repressing genes involved in cholesterol and fatty acid biosynthesis regulated by sterol response element binding proteins (SREBPs). TRC8 binds both SREBP-2 and SCAP (SREBP cleavage–activated protein) and hinders the transport of SREBP-2 from the ER to the Golgi apparatus for proteolytic processing, thereby reducing SREBP-2 target gene expression. 148 In a separate study, TRC8 induction destabilizes the precursor forms SREBP-1 and SREBP-2, presumably by promoting their ubiquitylation and consequent degradation by the proteasomes. 149 TRC8 also interacts with eIF3f and eIF3g, components of eIF3 in a larger translation initiation complex. TRC8 overexpression also inhibits protein translation. 149 TRC8 may function to coordinate lipid biosynthesis and protein translation during periods of cholesterol limitation.
RNF5/RMA1
RNF5 is an ER ubiquitin ligase originally found to associate with paxillin. 150 RNF5 ubiquitylates paxillin but does not target it for degradation. Rather, RNF5 catalyzes K63-linked polyubiquitin chains on paxillin, decreasing paxillin localization to focal adhesions. By regulating the localization of paxillin, RNF5 functions as a regulator of cancer cell motility. RNF5 is subsequently found to be predominantly localized to the ER and participates in ERAD. RNF5 promotes the degradation of mutant CFTR in conjunction with either gp78 or CHIP. 119,151 On the ER membrane, RNF5 associates with and ubiquitylates JAMP (JNK-associated membrane protein). This noncanonical ubiquitylation does not alter JAMP stability but functions to inhibit the recruitment of p97/VCP and proteasomes to the ER membranes for efficient ERAD prior to and following ER stress. 152 RNF5 expression is increased in breast cancer and related cell lines. 153 Suppression of RNF5 expression inhibits cell proliferation and caused a reorganization of the actin cytoskeleton in a p53-dependent manner. High levels of RNF5 are associated with poor outcome and metastasis in breast cancer, melanoma, and other cancers. 153 The roles of RNF5 in tumor progression are not well understood but may be related to its regulation of actin cytoskeleton or modulation of ER stress response.
Parkin
In addition to ER-resident ubiquitin ligases, some cytosolic ubiquitin ligases also function in mammalian ERAD. Parkin, which belongs to a family of ubiquitin ligases that include 2 RING fingers and a cysteine-rich In-Between-RING (IBR) region, has been identified as a ubiquitin ligase for Pael-R, a misfolded ER protein. 144
Parkin is commonly mutated in autosomal recessive juvenile Parkinsonism (AR-JP). Mitochondrial defects and ER stress have been implicated in Parkinson disease. Parkin is also a putative tumor suppressor gene located in the fragile site FRA6E. 154 Parkin protein is commonly lost in breast, ovarian, and lung cancer cell lines. 154 In addition, a large proportion (71%) of tumor tissues showed decreased or no expression of Parkin transcript relative to normal ovary or breast tissue. Frequent loss of heterozygosity and deletions spanning the Parkin gene are found in several cancers. 155-159 Ectopic expression of Parkin reduces cell growth and increases apoptosis in hepatocellular carcinomas, whereas expression of Parkin in lung cancer cells inhibits tumor formation in xenograft models. 157,159,160 Consistent with Parkin being a tumor suppressor, Parkin–/– mice are susceptible to development of hepatocarcinoma. 157 Parkin–/– mice show up-regulaton of follistatin, enhanced hepatocyte proliferation, and resistance to apoptosis in a follistatin-dependent manner. Follistatin is an autocrine glycoprotein and binds activin, inhibiting cellular proliferation while promoting cellular differentiation.
Parkin also induces the degradation of cyclin E, possibly as part of a SCF complex with the F-box protein Fbw7 (SCFFbw7). 161 Alternatively spliced variants of Parkin detected in colon cancer fail to degrade cyclin E, suggesting that loss of Parkin regulation of cyclin E contributes to colon cancer. 162
More recently, Parkin has been shown to play a role in mitophagy of damaged mitochondria. 163-170 Parkin translocates to depolarized mitochondria in a step that depends on Nix. 163 At the mitochondria, Parkin catalyzes the formation of K27- and K63-linked polyubiquitin chains. 164 VDAC1 is shown to be the target for Parkin K27-linked polyubiquitylation. Subsequent recruitment of p62/sequestosome-1 marks the mitochondria for autophagy. 164 Autophagic induction depends on Nix, which causes mitochondrial depolarization and mTOR inhibition. 163 AR-JP–linked Parkin mutations are defective in supporting mitophagy due to distinct defects at recognition, transportation, or ubiquitination of impaired mitochondria, thereby implicating mitophagy defects in the development of parkinsonism. Tumor cells often suppress their mitochondria in response to hypoxia and other stresses to reduce oxidative stress, a process that also utilizes autophagy. 171-174 Parkin’s role in mitophagy might also relate to its tumor suppressor functions.
The capacity of this cytosolic ubiquitin ligase to recognize a diverse set of substrates 175 may be related to Parkin’s interaction with the cytosolic chaperone HSP70 and ubiquitylate HSP70 client proteins. 176 The ability of Parkin to ubiquitylate HSP70 client proteins suggests that Parkin may play a role in the degradation of substrates normally stabilized by HSP90, many of which are important for tumor cell survival and proliferation. A reasonable working model for Parkin function may be that its activity towards substrates is largely regulated by the dynamics of HSP90 interaction with its client proteins. Understanding the roles of this chaperone-dependent mechanism in Parkin-mediated quality control could help unravel the mechanisms of Parkin in tumor suppression.
CHIP
CHIP is another cytosolic E3 that is implicated in ERAD. 177,178 CHIP is a U-box ubiquitin ligase that plays a role in protein quality control in collaboration with cytosolic chaperones. The U-box has similar tertiary structure as RING finger but does not bind zinc ions. Instead, salt bridges serve to maintain the integrity of the structure. CHIP has also been suggested to function together with Parkin in Pael-R degradation. 177 More recently, RNF5 and CHIP have been shown to function sequentially in recognizing the folding defect of the CFTR ΔF508 mutant and targeting this protein for degradation. 151
In a recent study, the levels of CHIP were found to correlate inversely with malignancy in breast tumors. 179 Consistent with a tumor suppressor function for CHIP, knockdown of CHIP increases growth of subcutaneous tumors. Conversely, overexpression of CHIP suppresses pulmonary metastasis of intravenously delivered breast cancer cells. This is largely due to CHIP degradation of the transcriptional coactivator SRC-3, resulting in reduced levels of Smad2 and Twist. 179 The F-box protein Fbw7, as well as E6AP, has previously been shown to down-regulate SRC-3 and suppress tumor growth. 180,181 Similar to Parkin, CHIP can ubiquitylate substrates bound to HSP70 and has been shown to regulate the degradation of proteins normally stabilized by chaperones. 182-186 It is not clear whether the effects of CHIP on SRC-3 depend on its role in chaperone-mediated degradation. The finding that CHIP suppresses tumorigenesis is rather surprising considering that CHIP also plays a role in the activation of heat shock response and recovery from stress, thereby increasing tumor cell survival. 187,188 Moreover, CHIP knockout mice develop aging phenotypes suggestive of senescence. 189 These observations suggest that certain cancer cells may have evoked mechanisms to evade normal regulation of CHIP.
Future Directions
Much remains to be discovered regarding the basic mechanisms of ubiquitylation and ERAD. Ubiquitylation has traditionally been considered as a modification of lysines in the substrate proteins. It was subsequently determined that the N-terminus of proteins can also be modified by ubiquitin. 190 An additional paradigm shift that has recently come about from studying ERAD with virally encoded E3s, 191-193 and more recently from studying Synoviolin regulation of TCRα, 194 is that ubiquitylation can occur on nucleophilic amino acids other than lysine, particularly in the absence of available lysine residues. Similarly, polyubiquitin chains built on the catalytic cysteine of Ubc7p can target this yeast E2 for degradation. 195 How ubiquitin ligases recognize their substrates and activate transfer of ubiquitin to the various acceptor residues on the substrates is an exciting area of research.
Increasingly, interests have focused on the roles of ER stress and the UPR in cancer. The tight coupling of ERAD and UPR suggests that targeting either of these sets of pathways can cause ER stress–induced cell death. Although this approach represents an attractive therapeutic strategy, it is plausible that cancer cells, which are constantly mounting mild UPR to cope with the stresses of cellular transformation, may continually develop mechanisms to evade cell death induced by ER stress. Actual changes in UPR and ERAD during cancer progression have not been carefully measured. Similarly, the roles of UPR and ERAD in cancer stem cells have not been explored.
Despite advances in cancer research, metastasis remains the major cause of cancer-related mortality. Much more remains to be learned about the roles of ER stress during cancer metastasis (Fig. 2). Studies in animal models have shown that initial survival and growth of tumor cells at the secondary site are major determinants of metastatic colonization of the site. While initial attrition of cells occurs by cell death mechanisms, a large number of cells can stay dormant for a long period of time only to initiate growth when the microenvironment is favorable. Cells may also stay dormant as micrometastases form in the balance of proliferation and apoptosis. Any condition that tips the balance towards proliferation can spur the growth of metastasis. It is therefore important to understand the factors that influence cell survival and proliferation during metastasis. The UPR and associated ERAD may be important factors that influence these processes. Metastasizing cells migrate from a hypoxic, nutrient-deprived, primary tumor to a new organ. In secondary sites such as the lungs, oxygen availability is initially not a limiting factor. However, it is reasonable that cells reaching the secondary sites secrete extracellular matrix and growth factors or cell surface receptors as they establish in the new environment. A functional ER response may be critical to the successful survival and initial growth of the cells. Improved imaging methods that detect UPR molecular events such as XBP1 or PERK activation in single cells could help address the role of ER stress during the early stages of metastasis. Since the UPR can have both prosurvival and cell death effects, understanding the temporal sequence of UPR activation during metastasis would be necessary to effectively target this system.
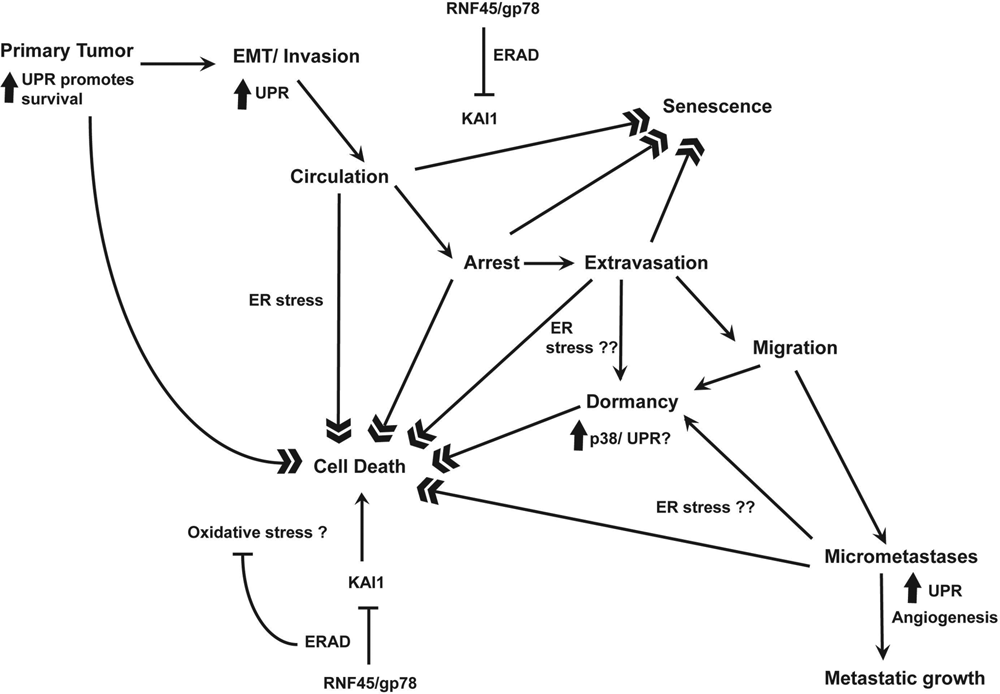
A model for studying the roles of UPR and ERAD in metastasis.
Finally, a critical question is whether ERAD and the UPR represent plausible targets for therapy. Most studies to date have deleted these genes before tumor development. Often, metastasis will be present when patients begin treatment. It will be important to know whether targeting the UPR and ERAD pathway can cause tumor regression after the metastases have already been established. As proliferating tumors and dormant tumors may be expected to respond differently to inhibition of ERAD and UPR, it would be important to understand the different roles of UPR in these tumors.
Footnotes
Acknowledgements
Work in the Weissman laboratory is supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, and Center for Cancer Research.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.