
Abstract
Members of the nuclear factor–κB (NF-κB) family of transcription factors play critical roles in regulating the expression of genes whose products are involved in inflammation, the immune response, cell proliferation, and the suppression of both death receptor– and stress-induced apoptosis. Abnormal NF-κB activation has been observed in various inflammatory diseases and many types of cancers. Gene knockout studies have clearly demonstrated that most of the physiologically relevant stimuli that activate NF-κB converge on the inhibitor of κB kinase (IKK). Although the mechanism by which IKK activates NF-κB is well established, the upstream signaling mechanisms—those that underlie IKK activation by IKK kinases (IKK-Ks)—are not yet fully understood. The current belief is that members of the TNF receptor–associated factor (TRAF) family function as ubiquitin E3 ligases that catalyze noncanonical polyubiquitination of adaptor proteins and that the ubiquitinated adaptor proteins in turn serve as platforms to recruit IKK and IKK-Ks, facilitating IKK activation through proximity-mediated phosphorylation. This review will focus on the most recent findings relating to the role of TRAF-mediated protein ubiquitination in regulating IKK activation and highlight the newly emerging complexity of protein ubiquitination in receptor-induced NF-κB activation.
Keywords
Introduction
The inducible regulation of gene expression is key to the ability of multicellular organisms to adapt to environmental changes and to cope with mechanical and chemical stresses. Soon after its identification (nearly 20 years ago) as a transcription factor (TF) that binds to the intronic enhancer of the κ light chain gene in B cells, nuclear factor–κB (NF-κB) emerged as a paradigm for the inducible TFs. 1 The evolutionarily conserved function of NF-κB is to regulate the innate and adaptive immune response, 2 and a large body of evidence gathered over the last 2 decades has revealed that many common diseases, including rheumatoid arthritis, diabetes, and cancer, are associated with NF-κB dysregulation. These discoveries have attracted broad interest, among investigators in the fields of immunology and tumor biology, in the signaling pathways that control NF-κB activity. 1,2
The NF-κB family of TFs consists of 5 members: p105/p50 (NF-κB1), p100/p52 (NF-κB2), p65 (RelA), c-Rel, and RelB. 2 All NF-κB family members contain an N-terminal Rel homology domain (RHD), which mediates DNA binding and homodimerization and heterodimerization. 2,3 Notably, only p65, c-Rel, and RelB harbor a transcription activation domain (TAD) necessary for the positive regulation of gene expression; as p50 and p52 lack this TAD, they may repress transcription when they are not associated with p65, c-Rel, or RelB. In unstimulated cells, NF-κB dimers are retained in the cytoplasm by a family of inhibitory proteins known as inhibitors of NF-κB (IκBs). 1,2 Mammalian cells express 3 classic IκBs (IκBα, IκBβ, and IκBϵ) and 2 novel IκBs (IκBζ and BCL-3). The IκB proteins are characterized by the presence of multiple ankyrin repeat domains that mediate IκB binding to NF-κB dimers and thereby interference with the nuclear translocation of the NF-κB dimers. The C-terminal halves of the p105 and p100 proteins, the precursors of p50 and p52, respectively, also harbor multiple ankyrin repeats. This allows them to serve an IκB-like function, retaining the partners with which they are paired in the cytoplasm. 2,3
Two Pathways, Canonical and Noncanonical, Activate NF-κB
NF-κB activation occurs mainly through 2 signaling pathways: the canonical and the noncanonical. 3 Activation of the canonical NF-κB pathway typically involves phosphorylation of IκBα by the IκB kinase (IKK) complex, which consists of the IKKα and IKKβ catalytic subunits and the NEMO (also known as IKKγ) regulatory subunit. 2,3 This phosphorylation leads to IκBα degradation via an ubiquitination-mediated and proteasome-dependent mechanism, allowing NF-κB (most commonly, the p65/p50 dimer) to translocate to the nucleus, bind DNA, and activate the expression of genes involved in inflammation and the innate immune response. This pathway is activated by a remarkably diverse array of stimuli, including proinflammatory cytokines, pathogen-associated molecules, growth factors, oxidative stress, and DNA damage. 3,4 Activation of the noncanonical NF-κB pathway, on the other hand, is dependent on the IKKα homodimer and independent of IKKβ and NEMO. 3 In this pathway, p100 is phosphorylated at 2 C-terminal sites by IKKα, and this causes p100 polyubiquitination. However, this polyubiquitination triggers degradation of only the C-terminal half of p100, releasing the N-terminal p52 portion. As the RHD of p100 is most commonly associated with RelB, activation of this pathway results in nuclear translocation of the RelB/p52 dimer and in the expression of genes involved in adaptive immunity and lymphoid organogenesis. 2,3 In contrast to the canonical pathway, the noncanonical NF-κB pathway is activated only by a subset of tumor necrosis factor (TNF) family members, such as lymphotoxin α/β (LTα/β), B cell–activating factor (BAFF), and the CD40 ligand (CD40L). 2,3
NF-κB Generally Promotes Tumor Formation and Progression
Epidemiological studies have clearly demonstrated that dysregulation of the inflammatory response as a consequence of infection and environmental insults is a major risk factor for various types of cancer. 4 For instance, chronic infection with hepatitis B or C viruses is a risk factor for hepatocellular carcinoma (HCC), chronic infection with Helicobacter pylori is associated with gastric cancer, and chronic airway inflammation caused by airborne particles and tobacco smoke is likely an important promoter of lung carcinogenesis. 5 It is now widely recognized that the NF-κB pathway serves as a mechanistic link between inflammation and cancer development. NF-κB is ubiquitously expressed and can be activated by almost all types of stimuli that cause inflammation and cellular stress, as a consequence of which it transactivates the expression of more than 200 genes, including proinflammatory cytokines (e.g., TNFα, interleukin-1 [IL-1], and IL-6), chemokines (e.g., IL-8/CXCL8), angiogenic factors (e.g., matrix metalloproteases and cyclooxygenase 2), adhesion molecules (e.g., intercellular adhesion molecule-I [ICAM-I]), and antiapoptotic proteins (e.g., cellular inhibitors of apoptosis 1 [cIAP1], cIAP2, cellular FLICE-inhibitory protein [cFLIP], and Bcl-XL). 4-6 NF-κB–induced cytokines in turn activate receptors that further propagate and amplify the inflammatory response, NF-κB–induced antiapoptotic proteins are the major determinants of the ability of neoplastic cells to resist apoptosis-based tumor-surveillance mechanisms, and NF-κB–induced chemokines and angiogenic factors promote inflammatory cell recruitment and angiogenesis, thereby promoting tumor formation, progression, and metastasis. 4-7 Indeed, gene knockout studies have provided solid evidence that NF-κB plays a causative role in malignant conversion and progression. Notably, selective inactivation of the Ikbkb gene (a gene encodes IKKβ) within enterocytes resulted in an 80% decrease in colitis-associated cancer induced by the procarcinogens azoxymethane and dextran-sulphate sodium. 8 Collectively, the literature suggests that NF-κB–induced gene products promote the transformation, survival, proliferation, metastasis, and chemoresistance of most types of cancer cells.
Although NF-κB has emerged as a critical promoter of inflammation-induced cancers, there is also evidence from mouse models of chemically induced skin and liver cancers that it can have the opposite effect. 4 For example, inhibiting NF-κB by targeted overexpression of IκBα superrepressor in hepatocytes led to a significant increase in the number of squamous cell carcinomas (SCCs) produced by exposure to the procarcinogen DMBA plus phorbol ester TPA. 9 Hepatocyte-specific deletion of the Ikbkb gene also greatly augmented the multiplicity and size of HCCs in mice treated with the procarcinogen diethylnitrosamine. 10 Notably, the increased tumor formation in these models seems to be associated with an increase in apoptosis and compensatory proliferation that resulted from prolonged activation of the c-Jun N-terminal kinase (JNK) in the absence of NF-κB activation. 8,11 JNK is a member of the mitogen-activated protein kinase (MAPK) family, which stimulates activator protein-1 (AP-1) transcription factors. 12 Recent gene knockout studies have revealed the existence of crosstalk between the JNK and NF-κB pathways. In normal cells, NF-κB inhibits the prolonged phase of JNK activation by inducing the expression of XIAP, Gadd45β, and cFLIP and also by suppressing the accumulation of reactive oxygen species (ROS). 13-15 As both hepatocytes and keratinocytes have strong regenerative capacity, the increase in carcinogenesis following inhibition of the NF-κB pathway in both of these models is likely due to enhanced local injury, increased ROS production, and prolonged JNK activation. 4 In fact, in the case of human HCC and SCC, it is unlikely that a decrease in NF-κB activity is responsible for increased tumorigenesis, as NF-κB is constitutively activated in both SCC and HCC cell lines and tumor specimens. 4,6 Overall, current knowledge suggests that NF-κB is a bona fide tumor promotor in most, although possibly not all, types of human cancers.
Although numerous studies have demonstrated that NF-κB is constitutively activated in various cancer types and that it plays a critical role in tumor progression, the mechanism underlying this constitutive activation remains poorly understood. 2,4 However, an increasing body of work has revealed that NF-κB activation in response to inflammatory cytokines and bacterial components is regulated by protein ubiquitination.
Two Forms of Protein Ubiquitination, Canonical and Noncanonical, Play Distinct Roles
Like protein phosphorylation, ubiquitination is an inducible and reversible process and is involved in almost every cellular process. 16,17 Protein ubiquitination occurs through sequential steps catalyzed by E1 (ubiquitin [Ub]–activating), E2 (Ub-conjugating; also known as Ubc), and E3 (Ub ligase) enzymes. E1 activates the Ub C-terminus in an ATP-dependent manner, leading to the formation of a thiolester bond between the C-terminal glycine of Ub and an active-site cysteine in the E1. Activated Ub is then transferred to an active-site cysteine of the E2, also forming a thiolester bond. Finally, E3 covalently attaches Ub to an amino group of lysyl residue(s) on the substrate protein. 16 After the Ub is linked to a substrate protein, a polyubiquitin (pUb) chain is usually formed, with the C-terminus of each Ub unit covalently linked to a specific lysyl residue of the previous Ub. Ub has 7 lysyl residues: K6, K11, K27, K29, K33, K48, and K63. A pUb chain linked via the K48 residue of Ub (K48-pUb) governs targeting of substrates to the 26S proteasome for degradation. This type of ubiquitination is the best characterized so far and is known as canonical protein ubiquitination. Recent proteomics and mass-spectrometry analyses revealed that, in fact, each of the other 6 lysine residues in Ub can also be used for pUb chain assembly. 18 Assembly of these non-K48-linked Ub chains is referred to as noncanonical ubiquitination. Whereas the substrate specificity of Ub ligation is determined by E3, the topology of the Ub chain seems to be controlled collaboratively by E2 and E3. 19 A special class of E2-like proteins is of particular interest in this context; these E2 variants retain the core Ub-conjugating (UBC) domain but lack the canonical active-site cysteine, and some E2 variants (e.g., Uev1A/MMS2) cooperate with E2s (e.g., Ubc13) to catalyze the assembly of K63-linked pUb (K63-pUb) chains. 17,19 The K63-pUb chain is of interest, as it has been implicated in a variety of nonproteolytic cellular functions. 16,17 The removal of Ub from substrates is carried out by deubiquitinating enzymes (DUBs), which rescue substrates from degradation, recycle Ub, and limit nonproteolytic functions of ubiquitination. With respect to the nonproteolytic functions of ubiquitination, the best characterized is activation of the NF-κB pathway. 20 In this review, I discuss the impact of ubiquitination and deubiquitination on the IL-1 receptor (IL-1R), TNF receptor 1 (TNFR1), and CD40 signaling pathways to illustrate the many ways in which this cycle controls NF-κB activation.
K63-Linked Ubiquitination of TRAF6 Links IL-1R Signaling to NF-κB Activation
Members of the IL-1R and Toll-like receptor (IL-1R/TLR) superfamily play crucial roles in innate and adaptive immunity. Most members of this superfamily activate NF-κB by similar mechanisms, which involve myeloid differentiation factor 88 (MyD88) and TNFR-associated factor 6 (TRAF6). 21,22 TRAF family proteins are characterized by the presence of a conserved C-terminal TRAF domain and, with the notable exception of TRAF1, an N-terminal RING finger domain. 23 The role of the K63-pUb chain in IL-1R–induced NF-κB activation was first reported by Chen and colleagues. 24 The current model regarding activation of NF-κB by IL-1R is that IL-1RI associates with IL-1R accessory protein (IL-1RAcP) following ligation with IL-1, after which these 2 receptors act together to recruit the adaptor protein MyD88 (Fig. 1). This leads to the further recruitment of IL-1R–associated kinase 1 (IRAK-1), IRAK-4, and the adaptor proteins Tollip and Pellino. Both IRAK1 and IRAK4 in turn recruit TRAF6, promoting its oligomerization and association with the E2 heterodimer, Ubc13/Uev1A. This interaction between TRAF6 and Ubc13/Uev1A induces RING-dependent autoubiquitination of TRAF6, through a K63 linkage. 20 The TRAF6-K63-pUb chain then recruits both the IKK complex and the transforming growth factor β–activated protein kinase 1 (TAK1) complex. 20,21 The TAK1 complex consists of TAK1, TAK1-binding protein 1 (TAB1), and either TAB2 or TAB3. TAB2 and TAB3 are homologous proteins that contain a highly conserved nuclear zinc finger (NZF)–type Ub-binding domain (UBD), and it is the UBD that allows the TAK1 complex to bind preferentially to K63-pUb chains. The NEMO protein of the IKK complex contains an N-terminal kinase–binding domain, 3 coiled-coil regions (CC1, CC2, and LZ), and a C-terminal zinc finger (ZF) domain. Deletion mapping experiments have revealed that the region encompassing the CC2 and LZ domains (also known as the UBAN domain for “Ub-binding domain in NEMO and ABIN”) can interact directly with K63-pUb chains. 25-27 Subsequent to the recruitment of the TAK1 and IKK complexes to the TRAF6-K63-pUb chain, TAK1 is activated, which in turn activates IKK by directly phosphorylating its activation loop. 20
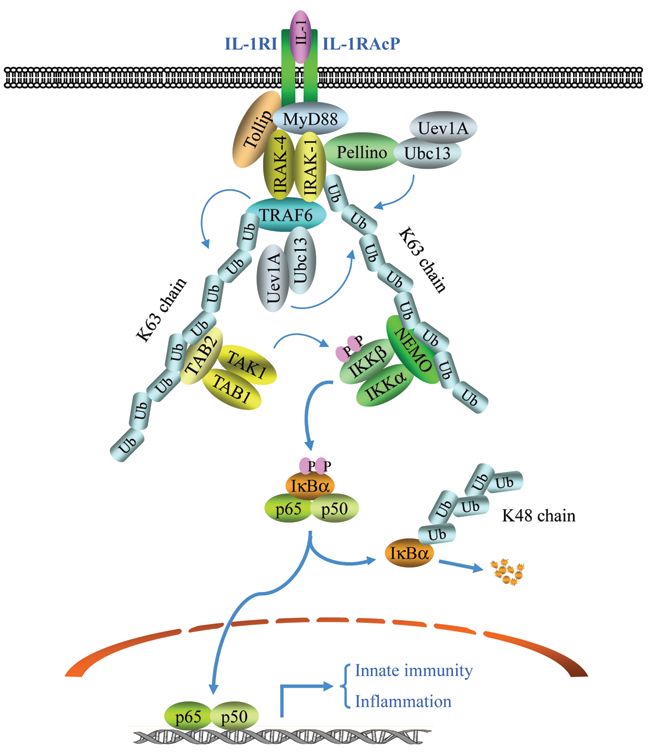
The canonical NF-κB activation by IL-1. Upon IL-1 stimulation, IL-1RI associates with IL-1RAcP and recruits MyD88, Tollip, IRAK-1, and IRAK-4, leading to IRAK1/4-dependent recruitment of TRAF6 and Pellino. TRAF6 then, in conjunction with Ubc13/Uev1A, undergoes K63-linked autoubiquitination, while Pellino catalyzes IRAK1 ubiquitination through K63 linkage. Ubiquitinated TRAF6 in turn serves as a platform to recruit the TAK1/TAB1/TAB2 complex, resulting in TAK1 activation. Simultaneously, ubiquitinated IRAK1 recruits the IKKα/IKKβ/NEMO complex, promoting TAK1-mediated activation of IKKβ. Activated IKKβ then directly phosphorylates IκBα, resulting in its K48-linked ubiquitination and degradation. This IκBα degradation allows canonical NF-κB (p65/p50 dimer) to translocate to the nucleus and activate the expression of genes involved in inflammation and innate immunity.
K63-Linked Ubiquitination of IRAK1 Also Links IL-1R Signaling to NF-κB Activation
Although numerous studies have demonstrated that Ubc13/Uev1A-mediated TRAF6 autoubiquitination is essential for IL-1R–induced activation of JNK and NF-κB, recent studies have revealed alternative pathways that can also lead to NF-κB activation. 28,29 The Ubc13/Uev1A heterodimer is the well-known E2 complex that specifically catalyzes the K63-linked ubiquitination of substrates. Surprisingly, conditional knockout of Ubc13 in B cells and MEFs severely impairs only JNK, and not NF-κB, activation in response to IL-1 stimulation. 29 As UbcH5 and UbcH7 have also been reported to mediate K63-linked ubiquitination, it is possible that some E2s are redundant for TRAF6 autoubiquitination. 7,19,20,30 However, Walsh et al. demonstrated that a TRAF6 mutant lacking all of its lysines (and thereby unubiquitinated) interacts with the TAK1-TAB1-TAB2 complex normally in vivo and that it is fully competent to rescue IL-1–mediated NF-κB and JNK activation in TRAF6-deficient MEFs. 28 Thus, additional substrates may be K63 ubiquitinated in response to IL-1 stimulation.
IRAK1 and IRAK4 are potent protein kinases and are activated immediately upon recruitment to the IL-1R. 31 The phosphorylation of IRAK1 has been reported to lead to its proteasome-dependent degradation, indicating that IRAK1 can undergo K48-linked ubiquitination. 31,32 However, recent studies revealed that IRAK1 undergoes K63-linked ubiquitination following IL-1 stimulation and that IRAK1 ubiquitination plays an essential role in the recruitment of NEMO and activation of IKK in this context. 33,34 Interestingly, NEMO preferentially bound IRAK1-K63-pUb over TRAF6-K63-pUb in immunoprecipitation experiments, even though both IRAK1 and TRAF6 were ubiquitinated in the cells following IL-1 stimulation. 33,34 Notably, in addition to TRAF6, the adaptor protein Pellino, which has a RING-like domain in the C-terminus, seems to function as an E3 ligase in the catalysis of K63-linked ubiquitination of IRAK1. 35 These studies partially explain why the lysine-deficient TRAF6 mutant can activate both the JNK and NF-κB pathways; that is, K63-linked polyubiquitination of TRAF6 and IRAK1 may have redundant functions in TAK1 activation, and both TRAF6 and Pellino may act as E3 ligases to catalyze K63-linked ubiquitination of IRAK1 (Fig. 1). However, many questions remain to be answered with regard to the role of other E2s in IL-1R–induced activation of JNK and NF-κB. For example, IL-1R–induced TRAF6 ubiquitination and TAK1 activation are normal in Ubc13-deficient cells, even though JNK activation is impaired, leaving in question whether TRAF6 autoubiquitination mediated by other E2s in Ubc13-deficient cells is K63 linked. Furthermore, it remains unclear why TRAF6 ubiquitination mediated by other E2s is able to activate NF-κB but not JNK.
IL-1 May Activate NF-κB Independent of K63-Linked Protein Ubiquitination
Certain data in the literature suggest the existence of alternative pathways that could lead to NF-κB activation in response to IL-1 stimulation independent of K63-linked ubiquitination. For example, Kobayashi et al. showed that the TRAF6 RING domain is essential for IL-1–induced activation of TAK1 and JNK, but not for IL-1–induced activation of NF-κB; specifically, reconstitution of TRAF6-deficient MEFs with wild-type or RING domain–deleted TRAF6 (TRAF6-WT or TRAF6-ΔR) at a physiological level led to a comparable level of NF-κB in the nucleus at 30 minutes after IL-1 stimulation. 36 Consistent with this finding, in NEMO-deficient cells reconstituted with a NEMO mutant (NEMO-L329P) that fails to interact with K63-pUb chains due to a point mutation in the UBAN domain, substantial IκBα degradation occurred by 15 minutes after IL-1 stimulation. 33 It appears that assessing NF-κB activation by gel mobility shift assay using a radioactive material is more sensitive than monitoring IκBα degradation by Western blotting. In light of the above-described studies, to better assess NF-κB activation, it might be wise to determine NF-κB activity by multiple different approaches. Nevertheless, the K63-pUb chain is likely to act as a platform to stabilize the receptor complex, resulting in efficient recruitment and rapid activation of the IKK complex to a level that could lead to almost complete degradation of IκBα within 5 minutes after stimulation. Although IKK might also be recruited to the receptors and activated to a certain degree in the absence of K63-linked ubiquitination, which would result in NF-κB nuclear accumulation, such IKK activation might not be sufficient to induce rapid and complete IκBα degradation.
RIP1 Ubiquitination Links TNFR1 Signaling to NF-κB Activation
Much of our current understanding of the signaling pathways that lead to NF-κB activation comes from studies of TNFR1 signaling. 1 The prevailing model in the field is that ligation of TNFR1 by TNFα induces the sequential recruitment of TNFR-associated death domain protein (TRADD), receptor inte- racting protein 1 (RIP1), and TRAF2/5 to TNFR1. Then, the RING domains of TRAF2 and/or TRAF5, in conjunction with Ubc13/Uev1A, catalyze K63-linked polyubiquitination of RIP1 at K377. This RIP1-K63-pUb chain in turn recruits both the TAK1/TAB1/TAB2 and IKKα/IKKβ̣/NEMO complexes by binding directly to the UBD domains present on TAB2 and NEMO, respectively. 20,25,26 Once “hooked” by RIP1-K63-pUb chains, TAK1 directly activates IKKβ through proximity-mediated phosphorylation (Fig. 2). 20,25,26 However, unlike TRAF6 whose E3 ligase activity has been demonstrated in in vitro ubiquitination assays by many independent investigators, the E3 ligase activities of TRAF2 and TRAF5 have not been unequivocally demonstrated in a completely purified system. 2 Nevertheless, TRAF2 is believed to have E3 ligase activity, and the RING domain–deleted TRAF2 (TRAF2-ΔR) has frequently been used as a dominant negative inhibitor of TNFα-induced activation of JNK and IKK in transient overexpression studies. 1,23 By expressing various RING-mutant forms of TRAF2 and carrying out siRNA-mediated knockdown of Ubc13, we have shown that both integrity of the TRAF2 RING domain and expression of Ubc13 are important for TNFα-induced activation of JNK, but not for that of IKK. 37 Recently, we also provided compelling evidence, by stably expressing TRAF2-ΔR in TRAF2 and TRAF5 double knockout (TRAF2/5 DKO) cells at a physiological level, that the TRAF2 RING domain plays a critical role in TNFα-induced JNK activation but that it is not essential for TNFα-induced IKK activation and NF-κB–dependent gene expression. 38 In support of our findings, Vince et al. recently reported that reconstituting TRAF2/5 DKO cells with TRAF2-ΔR completely restores TNFα-induced NF-κB activation. 39 These data suggest that other E3 ligases may be responsible for RIP1 ubiquitination in response to TNFα stimulation.
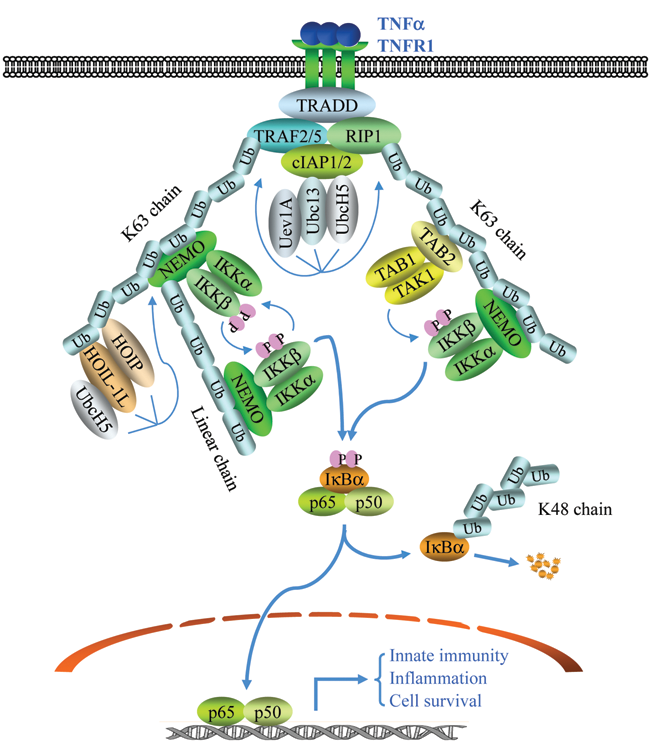
The TNFα signaling pathways to NF-κB activation. The binding of TNFα to TNFR1 leads to a rapid recruitment of TRADD, RIP1, TRAF2, TRAF5, cIAP1, and cIAP2. Formation of this complex triggers TRAF2/5 and cIAP1/2, in conjunction with UbcH5/Ubc13/Uev1A, to catalyze K63-linked polyubiquitination of RIP1 and autoubiquitination of TRAF2 and/or cIAP1 (not shown). K63 linkage–modified RIP1 then recruits the TAK1/TAB1/TAB2 and IKKα/IKKβ/NEMO complexes, leading to TAK1 activation and TAK1-mediated activation of IKKβ. Simultaneously, K63 linkage–modified TRAF2 and/or cIAP1 recruit the IKKα/IKKβ/NEMO and HOIL-1L/HOIP complexes, resulting in linear ubiquitination of NEMO. Such NEMO ubiquitination leads to more IKK recruitment, promoting further IKK activation via transautophosphorylation and also by stabilizing the TNFR1 complex. Activated IKK then phosphorylates IκBα, resulting in its K48-linked ubiquitination and degradation. This IκBα degradation allows p65/p50 dimer to translocate to the nucleus and activate the expression of genes involved in inflammation, innate immunity, and cell survival.
The TRAF2-cIAP1-Ubc13-UbcH5 Quaternary Complex Catalyzes RIP1 Ubiquitination
A growing body of studies has recently revealed that TRAF2 recruits cIAP1 and cIAP2 to the TNFR1 complex and that cIAP1/2 in turn catalyze RIP1 ubiquitination and NF-κB activation. 30,40,41 Although genetic deletion of either cIAP1 or cIAP2 had no effect on TNFα-induced NF-κB activation, siRNA-mediated knockdown of both resulted in significantly attenuated RIP1 ubiquitination and NF-κB activation. 30,41 Consequently, cells missing both cIAP1 and cIAP2 were sensitized to TNFα-induced apoptosis. 30,41 Bertrand et al. have shown in an in vitro ubiquitination assay that both cIAP1 and cIAP2 can act in conjunction with Ubc13/Uev1A to catalyze K48- and K63-linked RIP1 ubiquitination, in spite of the fact that Ubc13/Uev1A is considered to specialize in generating K63-pUb chains. 40 On the other hand, Varfolomeev et al. have shown, also using an in vitro ubiquitination assay, that cIAP1/2 collaborate with UbcH5, but not with Ubc13, to catalyze K48- and K63-linked polyubiquitination of RIP1. 30 In spite of the discrepancies between these studies regarding which E2s are involved in RIP1 ubiquitination, the analyses are consistent in suggesting that cIAPs can catalyze K48- as well as K63-linked polyubiquitination of RIP1 in vitro. It needs to be mentioned here that cIAPs are known to associate with Ubc4/5 and to target their substrates for degradation by catalyzing K48-linked ubiquitination. 7,42 In fact, cIAP1 knockout (KO) cells express markedly elevated levels of cIAP2 protein in the absence of increased mRNA expression. 43 In addition, TNFR2-mediated degradation of TRAF2 and ASK1 is severely impaired in cIAP1-deficient B cells. 44 Therefore, there is no doubt that cIAP1 targets some of its substrates for degradation in certain settings. However, TRAF2 is not degraded in the context of TNFα-triggered TNFR1 signaling, although both TRAF2 and cIAP1 are recruited to the TNFR1 complex. This suggests that cIAP1 may catalyze nondegradative protein ubiquitination within the TNFR1 complex. 2,23
The RING domains of IAPs reportedly bind to Ubc4, UbcH5, Ubc6, Ubc7, Ubc8, and Ubc13/Uev1A. 7,42 UbcH5 seems more promiscuous and can promote the ubiquitination of a surprisingly diverse array of products, even synthesizing chains of mixed linkages. 19,45 In addition, UbcH5 is highly active and efficient in targeting its substrates for ubiquitination. 45 Ubc13/Uev1A, on the other hand, which appears to be very efficient at forming K63-linked chains, nevertheless performs poorly in attaching the first Ub moiety to a target substrate. 19,45 Windheim et al. have recently shown that Ubc13/Uev1A-catalyzed K63-pUb chains are not at all attached to any substrates. 46 It appears that conjugation of pUb chains of a particular topology requires the cooperative actions of 2 different classes of E2s. 47 For example, although UbcH5 readily conjugates the first Ub to a lysine residue in the substrate, this enzyme lacks specificity for any particular type of chain elongation subsequently. Therefore, the assembly of K63-pUb chains on a target substrate appears to be the result of a cooperative interaction between chain-initiating UbcH5 and chain-elongating Ubc13/Uev1A. 19 Recently, Xu et al. have demonstrated that IKK activation by TNFα also requires UbcH5, which in collaboration with cIAP1 catalyzes polyubiquitination of RIP1 not restricted to the K63-pUb chain. 48 The recruitment of cIAP1 to TNFR1 is dependent on TRAF2, and many investigators have shown independently—using Ub mutants, siRNA-mediated knockdown of Ubc13, and an anti-K63-pUb-selective antibody—that Ubc13 contributes to K63-linked ubiquitination of RIP1. Thus, it is most likely that TRAF2, cIAP1, UbcH5, and Ubc13 function cooperatively to catalyze the assembly of the K63-pUb chain on RIP1 (Fig. 2). This notion is supported by the fact that 1) RIP1 ubiquitination is impaired in TRAF2/5 DKO cells as well as in cIAP1/2 knockdown cells; 2) knockdown of either UbcH5 or Ubc13 attenuates RIP1 ubiquitination; and 3) reconstitution of TRAF2/5 DKO cells with a TRAF2 mutant that has an intact RING domain but cannot bind cIAPs fails to restore RIP1 ubiquitination. 20,25,26,38,40,48
TNFα Can Activate NF-κB in the Absence of TRAF2/5 Expression and RIP1 Ubiquitination
TRAF2 contributes to both canonical and noncanonical NF-κB activation. 2,3 Gene knockout studies have revealed that TRAF2 loss impairs TNFα-induced JNK activation without affecting IKK activation. 49 Tada et al. have reported that TRAF2/5 DKO MEFs exhibit an almost complete loss of TNFα-induced NF-κB activation. 50 These studies suggest that of TRAF2 and TRAF5, only TRAF2 has a role in JNK activation but that the 2 proteins have redundant roles in TNFα-triggered IKK activation. Surprisingly, the generation of conditional knockout mice using the Cre-loxP recombination system revealed that TRAF2 ablation in the B cell lineage results in constitutive activation of the noncanonical NF-κB pathway. 51 Subsequent analysis at the molecular level revealed that the constitutive activation of noncanonical NF-κB in TRAF2-deficient cells is due to an accumulation of NIK, which is otherwise constitutively degraded by nonredundant and cooperative actions of TRAF2/3 and cIAP1/2 (Fig. 3A). 52,53 A subset of TNFR members, including LTβR, CD40, and BAFFR, activates both the canonical and noncanonical NF-κB pathways. 3 It is believed that these receptors induce IκBα degradation in a NIK-dependent manner, as expression of a dominant-negative NIK (DN-NIK) inhibits IκBα degradation induced by LTα/β, CD40L, and BAFF. 54-56 In a majority of primary myeloma patient samples and cell lines, NIK is overexpressed due to genetic or epigenetic alterations in genes (e.g., Traf2, Traf3, Cyld, Bir2, and Bir3) associated with the regulation of NIK protein stability. 54,57 An accumulation of NIK in these multiple myeloma cell lines results in constitutive activation of both NF-κB pathways, as siRNA-mediated knockdown of NIK in these cell lines affects signaling by both, decreasing phosphorylation of IκBα and the abundance of nuclear p52. 54,57 All these studies suggest that an accumulation of NIK leads to constitutive activation of both NF-κB pathways and that the canonical NF-κB pathway may be constitutively activated in TRAF2/5 DKO cells, as NIK accumulates in these cells as a consequence of a lack of TRAF2 expression.
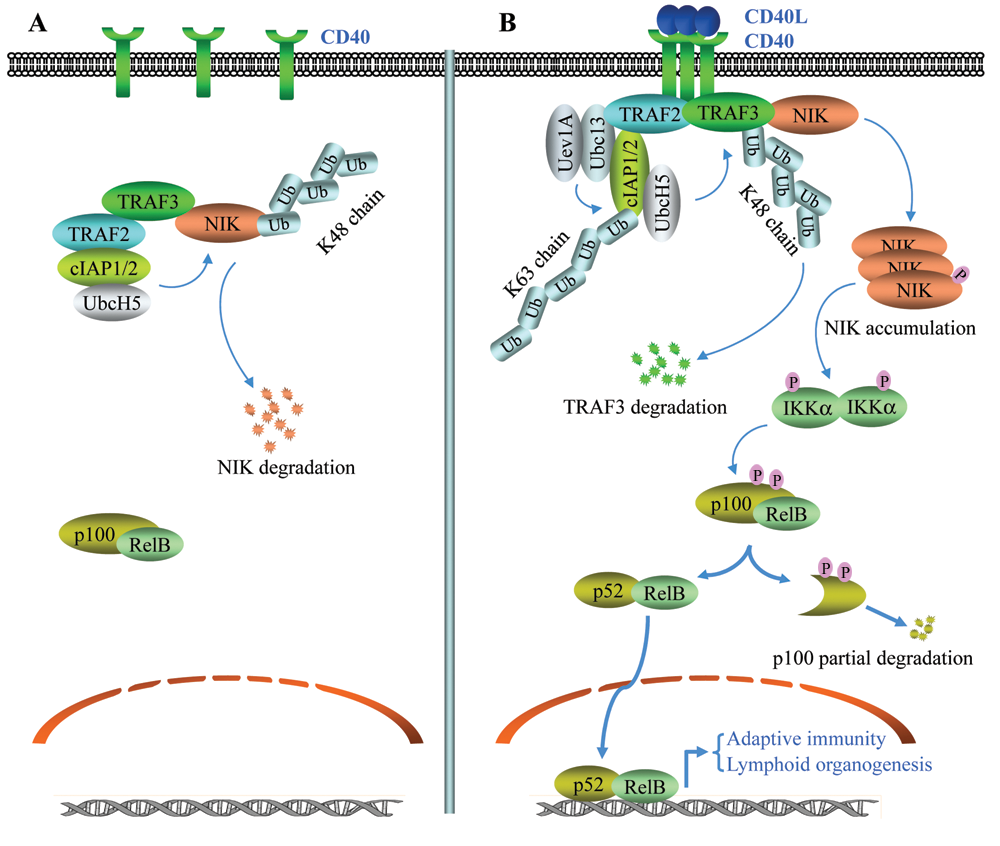
The noncanonical NF-κB activation by CD40L. (
In the NF-κB field, it is well accepted that simultaneous knockout of TRAF2 and TRAF5 abolishes TNFα-induced activation of canonical NF-κB. 2,4,50 However, this conclusion is based on an analysis of IκBα protein levels in TRAF2/5 DKO cells. 50 Recently, we reported that in both TRAF2 KO and TRAF2/5 DKO MEFs, the canonical IKK complex is constitutively activated to a certain level due to the accumulation of NIK and that stimulating these cells with TNFα further increases IKK activity in the absence of RIP1 polyubiquitination. 58 Notably, in TRAF2/5 DKO cells, constitutively activated IKK leads to constitutive IκBα phosphorylation, degradation, and resynthesis, effectively masking the immediate and complete degradation of IκBα induced by TNFα. 58 Therefore, the failure to observe complete IκBα degradation in TRAF2/5 DKO cells following TNFα exposure is not a consequence of impaired IKK activation but rather of constitutive degradation and resynthesis of IκBα prior to stimulation, as the Ikba gene itself is a target of NF-κB. We also found that the expression of DN-NIK or application of a NIK-targeting siRNA in TRAF2/5 DKO cells reduces basal IKK activity and significantly increases TNFα-induced immediate IKK activation. 58 In addition, 3 groups recently independently generated TRADD knockout mice and found that TNFα-induced activation of JNK and IKK is severely reduced in TRADD-deficient MEFs, due to the impaired recruitment of RIP1 and TRAF2 to the TNFR1 complex. 59-61 In TRADD-deficient macrophages, in contrast, RIP1 is directly recruited to TNFR1 in response to TNFα stimulation due to its high level of expression yet is not ubiquitinated because TRAF2 is not recruited in the absence of TRADD. Surprisingly, RIP1 recruitment to TNFR1 in this context is sufficient to activate IKK in the absence of K63-linked RIP1 polyubiquitination. 60,61 In addition, Xu et al. have surprisingly shown, using an Ub replacement strategy, that neither the K63-pUb chain nor the catalytic activity of Ubc13 is required for TNFα-induced IKK activation. 48 Even more surprisingly, Wong et al. have recently reported that TNFα can normally activate NF-κB in both primary and immortalized RIP1 KO MEFs, as well as in several cell lines isolated from E18 RIP1 KO embryos. 62 Collectively, these data suggest the existence of an alternative pathway that can mediate TNFα-induced IKK activation in the absence of TRAF2/5 expression and RIP1 ubiquitination. It has been shown that RIP1 recruits IKK to the TNFR1 complex in TRAF2-deficient cells by directly interacting with NEMO following TNFα stimulation and that TRAF2 recruits IKK in RIP1-deficient cells by directly interacting with IKKα and IKKβ. 58,63-65 Therefore, it is likely that TRAF2 and RIP1 independently recruit IKK to TNFR1 and that they can independently induce IKK activation by transautophosphorylation.
TAK1 Is Redundant with Other IKK-Ks in the NF-κB Pathway
Although TAK1 ablation severely affects the activation of both JNK and NF-κB in response to IL-1 and TNFα exposure, TAK1-deficient B cells display normal NF-κB but impaired JNK activation in response to the ligation of the B cell receptor. 66,67 In addition, whereas NF-κB activation is only moderately reduced in TAK1-deficient MEFs stimulated with LPS, JNK activation is almost completely impaired in this context. 66,67 These data suggest that as yet uncharacterized TAK1-independent pathways capable of activating NF-κB exist. One of the candidate kinases is mitogen-activated protein kinase kinase kinase 3 (MEKK3), as IL-1–, TNFα-, and LPS-induced NF-κB activation is also severely reduced in MEKK3-deficient MEFs. 68,69 It is worth noting that TLR8-mediated JNK and NF-κB activation is almost completely abolished in MEKK3-deficient MEFs, whereas it is unaffected in TAK1-deficient MEFs. 70 Interestingly, Blonska et al. have shown that reconstituting RIP1-deficient Jurkat T cells with a fusion protein composed of full-length MEKK3 and the death domain (DD) of RIP1 (MEKK3-DD) fully restores TNFα-induced NF-κB activation. 71 In contrast, stable expression of a fusion protein composed of NEMO and the RIP1 DD (NEMO-DD) cannot restore TNFα-induced NF-κB activation in RIP1-deficient cells, although NEMO-DD is also recruited to the TNFR1 complex in response to TNFα stimulation in these cells. 71 These data suggest that RIP1-mediated recruitment of MEKK3 to TNFR1 plays a specific role in activating IKK in response to TNFα stimulation and that forced recruitment of NEMO to the TNFR1 complex is insufficient for NF-κB activation. 71 It is most likely that both TAK1 and MEKK3 function as IKK-Ks to activate IKK and that TAK1 plays a more important role in JNK activation than in NF-κB activation following the ligation of certain inflammatory receptors.
Linear Ubiquitination of NEMO Promotes NF-κB Activation
Adding to the already existing complexity of K63-linked ubiquitination, linear ubiquitination has emerged as a new player in TNFR1-induced NF-κB activation. At present, the only known E3 ligase that specifically catalyzes linear ubiquitination is the linear Ub chain assembly complex (LUBAC), and the only known substrate modified by the linear Ub chain is NEMO. 72 LUBAC consists of 2 RING finger proteins, HOIL-1L and HOIP, and possesses a unique feature that allows it to assemble a head-to-tail linear pUb chain. 73 Intriguingly, LUBAC can use multiple E2s, including UbcH5, E2-25, and UbcH7, to generate head-to-tail Ub conjugates, suggesting that LUBAC, rather than the associated E2, is responsible for determining the linkage type. 73 LUBAC seems to bind to Ub chains of different linkage types, and its recruitment to the TNFR1 complex is dependent on TRAF2-cIAP1–mediated ubiquitination of TNFR1-associated effectors. 74 Surprisingly, although LUBAC reportedly binds to NEMO, and RIP1 is the most prominent target of TRAF2-cIAP1–mediated ubiquitination, the recruitment of LUBAC to TNFR1 is not affected in NEMO- and RIP1-deficient cells. 74 This may be because TRAF2 and cIAP1 are also ubiquitinated following TNFα stimulation. Once recruited to the TNFR1 complex, LUBAC can conjugate linear Ub chains onto specific lysine residues in the UBAN domain of NEMO, and this ubiquitination leads to stabilization of the TNFR1 complex. Thus, LUBAC-mediated linear pUb chains act as additional scaffolds that protect the TNFR1 complex from falling apart. 74 The UBAN domain of NEMO, which was thought to specifically bind K63-pUb chains, has been reported to display a 100-fold greater affinity for linear Ub chains. 75 This suggests that linear chains on NEMO can be recognized by UBAN motifs in neighboring NEMO molecules and that NEMO ubiquitination thus facilitates the recruitment of the additional IKK complexes (Fig. 2). 74 One caveat to this theory is that in HOIL-1L–deficient cells, TNFα-induced IκBα degradation is not fully impaired. 76 In addition, in the case of IL-1R signaling, NEMO ubiquitination is impaired in Ubc13-deficient cells following IL-1 stimulation, but IKK activation is normal. 36 Therefore, it is likely that linear ubiquitination of NEMO enhances IKK activity but is not essential for initial IKK activation.
Two Deubiquitinases, A20 and CYLD, Negatively Regulate NF-κB Activation
A20 was identified in a yeast 2-hybrid screen as a TRAF2-interacting protein that negatively regulates the NF-κB pathway. 77 Polymorphisms in Tnfaip3 (the A20-encoding gene) are associated with several autoimmune disorders, including Crohn disease, rheumatoid arthritis, and systemic lupus erythematosus. 77 Recent studies have demonstrated that Tnfaip3 is inactivated by mutation and/or promoter methylation in several lymphomas, including marginal zone lymphoma, diffuse large B cell lymphoma, follicular lymphoma, Burkitt lymphoma, and Hodgkin lymphoma. 77 Functional studies revealed that A20 is a dual-function enzyme, acting as a deubiquitinase as well as an E3 ligase in the cytokine-induced canonical NF-κB pathway upstream of the IKK complex. 78,79 At its N-terminus, A20 has an ovarian tumor (OTU) domain with deubiquitinating function toward K63-pUb, and at its C-terminus, it has 7 zinc-finger motifs possessing an E3 ligase activity capable of catalyzing K48-pUb chain assembly. 79 In TNFR1 signaling, RIP1-K63-pUb chains are recognized by A20, cleaved via the deubiquitinase activity of this enzyme, and replaced by K48-pUb chains. In this way, RIP1 is targeted for degradation. In IL-1R signaling, on the other hand, A20 cleaves K63-pUb chains attached to TRAF6 and IRAK1 and then targets them for K48-linked ubiquitination and degradation. 77,78 As a result, A20 limits the duration of NF-κB activation by the so-called Ub-editing mechanism. 78
Notably, recent studies have revealed that the regulation of A20 function is more complex than previously thought. Specifically, it appears that the removal of K63-pUb from RIP1 by A20 requires 3 additional proteins: Tax1-binding protein 1 (TAX1BP1), Itch, and RNF11. 80-83 TAX1BP1 has an Ub-binding ZF domain, which seems essential for the recruitment of A20 to RIP1-K63-pUb and TRAF6-K63-pUb. Thus, TAX1BP1 may serve as a bridge between A20 and its substrate. 80,81 In TAX1BP1-deficient cells, A20 fails to deubiquitinate RIP1-K63-pUb and TRAF6-K63-pUb, resulting in persistence of the TNFα- and IL-1–triggered IKK and JNK activation. 81 Itch is a HECT domain E3 ligase and binds to TAX1BP1 through its WW domain. In Itch-deficient cells, A20 also fails to deubiquitinate RIP1-K63-pUb, and this results in persistent NF-κB activation following TNFα stimulation. Thus, Itch also appears to be required for the association of A20 with ubiquitinated RIP1. 82 RNF11 has a RING finger E3 ligase activity, and immunoprecipitation experiments have revealed that endogenous RNF11 interacts with A20, TAX1BP1, and RIP1 following TNFα stimulation. 83 siRNA-mediated RNF11 knockdown in human THP-1 monocytes resulted in prolonged NF-κB and JNK activation in response to TNFα stimulation, due to increased RIP1 polyubiquitination. Furthermore, immunoprecipitation experiments indicate that RNF11 is required for the association between A20 and RIP1 that takes place in response to TNFα stimulation. 83 These data suggest that TAX1BP1, Itch, and RNF11 all facilitate substrate deubiquitination by A20. However, the role of TAX1BP1 in facilitating A20’s deubiquitinase activity may be restricted to certain tissues, as TAX1BP-deficient mice develop only inflammatory cardiac valvulitis, whereas A20-deficient mice exhibit systemic inflammation. 80,84 Furthermore, as recombinant A20 can directly ubiquitinate purified RIP1 in vitro, it is not clear whether Itch and RNF11 also function as E3 ligases to catalyze RIP1 ubiquitination or merely act as adaptor proteins to recruit A20 to its substrates. Further studies are needed to clarify how 3 E3 ligases (A20, Itch, and RNF11) cooperatively downregulate NF-κB and JNK activation by an Ub-editing mechanism.
CYLD was identified as a tumor-suppressor gene that is mutated in familial cylindromatosis, 85 and reduction in and/or absence of CYLD expression have also been observed in a variety of human tumor types including skin, kidney, liver, and uterine cervix. 86 CYLD contains a C-terminal Ub hydrolase domain and exhibits a deubiquitinating function. 86 In an siRNA screen searching for deubiquitinases that negatively regulate the NF-κB pathways, CYLD was identified as a deubiquitinase that suppresses NF-κB activity. 87 In yeast 2-hybrid screens using NEMO as bait, 2 groups independently identified CYLD as a protein that specifically interacts with NEMO. 88,89 Subsequent functional studies revealed that CYLD specifically removes K63-pUb chains from several mediators of the canonical NF-κB pathway, including TRAF2, TRAF6, and NEMO. 87-89 However, unlike A20 deficiency, which results in systemic inflammation and premature lethality, CYLD deficiency does not cause an apparent phenotype, not even a change in life-span. 90 However, the Cyld −/− mice are sensitized to the development of skin tumors; when they were subjected to a standard 2-stage (DMBA/TPA or DMBA/UVB) skin tumor induction protocol, they developed more and larger skin tumors than their wild-type (WT) counterparts, suggesting that CYLD suppresses the induction of skin tumors by environmental factors. Notably, this increase in the development of skin tumors in Cyld −/− mice seems not to depend on increased activation of canonical NF-κB, as neither UVB nor TPA was able to efficiently activate canonical NF-κB in keratinocytes; instead, it is dependent on increased ubiquitination and nuclear accumulation of BCL3. BCL3 is referred to as a noninhibitory member of the IκB family, as it functions as a transcriptional coactivator of NF-κB, specifically of the p50/p50 and p52/p52 homodimer. 2,86 K63-linked BCL3 ubiquitination appears to induce its translocation from the cytoplasm to the nucleus, where it associates with p50/p50 and p52/p52, converting them from transcriptional repressors to transcriptional activators, and thereby elevating the expression of proliferation-promoting target genes such as cyclin D1. 86,90 How K63-linked ubiquitination promotes BCL3 nuclear accumulation, however, has not been resolved.
It is worthy of mention that of all the deubiquitinases, only CYLD seems to efficiently cleave linear Ub chains. 91 As both A20 and CYLD are ubiquitously expressed, it is possible that they negatively regulate NF-κB by deubiquitinating different substrates in the NF-κB pathway, with A20 removing K63-linked chains from RIP1 and TRAF6, and CYLD removing linear Ub chains from NEMO. Further studies are needed to clarify physiological substrates of CYLD and A20 and to elucidate how these 2 deubiquitinases selectively target different substrates.
TRAF3 Degradation Links CD40 Signaling to Noncanonical NF-κB Activation
CD40 is expressed throughout B cell development and has important roles in B cell proliferation, differentiation, and immunoglobulin isotype switching. 92 CD40 activates both the canonical and noncanonical NF-κB pathways. The cytoplasmic tail of CD40 contains overlapping TRAF2- and TRAF3-binding sites at the C-terminus and a separate TRAF6-binding site at the membrane-proximal region. Mutation of these TRAF-binding sites revealed that each independently contributes to canonical NF-κB activation. 93 Overexpression of DN-NIK inhibited TRAF2/3-mediated, but not TRAF6-mediated, NF-κB activation in response to CD40 ligation, suggesting that TRAF2/3 may activate the canonical NF-κB pathway through NIK. 93 CD40 activation has been shown to induce rapid degradation of TRAF2 and TRAF3, but not that of TRAF6. 92,94 Degradation of TRAF2 and TRAF3 is preceded by a form of ubiquitination that requires an intact TRAF2 RING domain, as TRAF2-ΔR neither undergoes self-ubiquitination nor is able to induce TRAF3 ubiquitination following CD40 ligation. 95 When CD40-induced TRAF2/3 degradation was inhibited by blocking proteasomal activity, CD40-mediated canonical NF-κB activation was amplified and sustained, suggesting that TRAF2/3 degradation limits CD40 signaling to canonical NF-κB activation. 92,96 An Epstein-Barr virus (EBV)–encoded oncogenic protein, latent membrane protein 1 (LMP1), binds TRAF2/3 and mimics CD40 signaling; notably, LMP1 fails to cause TRAF2/3 degradation, and thus, its activation of the canonical NF-κB pathway is constitutive. 92,94
On the other hand, recent gene knockout studies have revealed that ablating either TRAF2 or TRAF3 results in constitutive activation of noncanonical NF-κB. 51,97 Notably, in cIAP1 or cIAP2 single-knockout cells, the noncanonical NF-κB pathway is normal and not constitutively activated, whereas in cells in which both cIAPs are depleted by application of an siRNA or IAP antagonists, it is constitutively activated. 7,98 We have demonstrated that stable expression of TRAF2-WT, but not of TRAF2-ΔR, in TRAF2/5 DKO cells suppresses NIK accumulation and constitutive p100 processing. 38 Similarly, He et al. have reported that the RING domain of TRAF3 must be structurally intact to inhibit noncanonical NF-κB in resting cells. 99 Most recently, Feltham et al. generated cIAP1/2 DKO mice and demonstrated that the RING domain of either cIAP1 or cIAP2 is required for suppressing p100 processing. 100 These studies suggest that TRAF2 and TRAF3 play nonredundant and complementary functions, whereas cIAP1 and cIAP2 play a redundant role in targeting NIK for degradation (Fig. 3A). In the case of noncanonical NF-κB activation in response to CD40 ligation, it appears that CD40 engagement induces the assembly of a complex, through TRAF3-mediated recruitment of NIK and TRAF2-mediated recruitment of cIAP1/2, to the receptor. Within this complex, TRAF2 activates cIAP1/2 by attaching K63-pUb chains, which in turn triggers cIAP1/2-mediated K48 ubiquitination of TRAF3. This results in TRAF3 degradation and NIK accumulation (Fig. 3B). 52,101,102 With respect to JNK activation, it appears that cIAP1/2-UbcH5–mediated TRAF3 degradation and TRAF2-Ubc13–mediated MEKK1 K63 ubiquitination trigger internalization of the receptor signaling complex to the cytoplasm, where TAK1 and MEKK1 activate the JNK pathway. 102 However, these studies raised new questions about whether TRAF2-cIAP1–mediated ubiquitination of effectors within the CD40 signaling complex also contributes to canonical NF-κB activation. It seems that neither Ubc13 expression nor TAK1 activation is required for CD40-induced IKKβ activation. 102 CD40 induces immediate and transient activation of IKKβ, which occurs well before NIK stabilization in response to CD40 ligation. It is possible that the recruitment of IKK to CD40 leads to its activation via transautophosphorylation and that TAK1 is responsible for amplifying the IKK signal once the initial activation step has occurred. 1,102 However, it is not clear how the IKK complex is recruited to the CD40 complex and how the TRAF2-cIAP1-UbcH5-Ubc13 complex catalyzes K48 ubiquitination of TRAF3 and K63 ubiquitination of MEKK1 following CD40 ligation.
Conclusions and Future Perspectives
The evasion of apoptosis is one of the common hallmarks of all types of cancer cells. 103 Although a tremendous number of studies have demonstrated that NF-κB is constitutively activated in various cancer types and that NF-κB plays a critical role in preventing cancer cells from death receptor– and stress-induced apoptosis, the mechanism underlying the constitutive activation of NF-κB in cancer cells remains poorly understood. 2,4 Gene alterations that could account for this (e.g., IκBα mutation in Hodgkin lymphoma, IKKϵ amplification in breast cancer tissue, and elevated NIK expression in multiple myeloma) have been reported. However, the frequency of such events is extremely low compared to the frequency of constitutive NF-κB activation in many types of human cancer. 6,57,104,105 Gene-targeting studies have demonstrated that the IKK complex is a point of convergence for NF-κB activation by a large number of stimuli. Notably, although the molecular mechanism by which IKK activates NF-κB has become clear, the mechanism by which IKK itself is activated by various stimuli still has not been fully elucidated. 1,2
The activation of IKK in response to inflammatory cytokines and bacterial components is believed to be regulated by K63-linked ubiquitination of effectors such as TRAF6 and RIP1. However, as discussed above, recent studies have indicated that either K63-linked ubiquitination of TRAF6 and RIP1 is not essential for NF-κB activation or at least one alternative pathway can lead to NF-κB activation. In addition, accumulating information is revealing that the regulation of K63-linked ubiquitination is more complicated than previously thought. For example, in TNFR1 signaling, the TRAF2-cIAP1-Ubc13-UbcH5 quaternary complex catalyzes K63-linked ubiquitination of RIP1, whereas in CD40 signaling, the same complex catalyzes K48-linked ubiquitination of TRAF3. Moreover, it appears that K63-pUb–modified substrates can also be efficiently targeted for proteasome-mediated degradation both in vitro and in vivo. 106 These studies have raised more questions than answers with regard to the role of protein ubiquitination in IKK activation. First, how does the same TRAF2-cIAP1 complex conjugate Ub to RIP1 and TRAF3 by different linkage? Second, how is the stability of K63-pUb–modified substrates regulated in vivo? Third, what type of Ub linkage is established on RIP1 by the TRAF2-cIAP1 complex under physiological conditions? Fourth, are any components of the TNFR1 complex other than RIP1 ubiquitinated via K63 linkage by the TRAF2-cIAP1 complex? Fifth, what are the alternative pathways that mediate TNFα-induced IKK activation in TRADD- and TRAF2/5-deficient cells in the absence of RIP1 ubiquitination? Clearly, much more needs to be understood about how the particular topology of the Ub linkage and the stability of K63-pUb–modified substrates are regulated. Recent studies suggest that 2 E2s cooperatively catalyze K63-pUb assembly and that Ub-binding proteins (UBP) play a critical role in propagating signals to the downstream effectors. Future studies applying live-imaging techniques to visualize Ub signaling networks in vivo, as well as proteomic approaches to study the dynamics of Ub-UBP interactions upon ligand stimulation, will make possible a more comprehensive analysis of cellular signaling events that are governed by ubiquitination.
Footnotes
Acknowledgements
The author apologizes to all scientists whose contributions to the field are not cited owing to space restriction. The author also thanks the members of the laboratory for critical reading of the paper.
Support by National Cancer Institute (NCI) grant CA78419 is gratefully acknowledged.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.