
Abstract
Chromosome 11 aberrations constitute the second most frequent chromosomal aberration in mouse plasmacytomas (PCTs) in which both the myc and abl oncogenes are constitutively expressed. In these tumors, previous G-banding studies had revealed numerical aberrations including duplication of the entire chromosome 11 or segments of telomeric bands D and E. The trisomy of chromosome 11 was always associated with accelerated pristane + v-abl/myc–induced PCT development. In the present study, PCT development was studied in a unique BALB/c congenic mouse strain, (T38HxBALB/c) F1, carrying a reciprocal translocation between chromosomes X and 11. After v-abl/myc induction, PCTs in this strain had acquired a nonrandom duplication of subcytoband 11E2. This duplication was always associated with accelerated PCT development. Corresponding synteny regions in the human and rat are changed in many tumors and involved in duplication, amplification, or translocation events. Thus, together with these synteny data, our findings strongly suggest a causal involvement of 11E2 in the acceleration of v-abl/myc–induced PCTs.
Introduction
Historically, mouse plasmacytoma, rat immunocytoma, and human Burkitt lymphoma were the first B lineage tumors shown to result from c-myc–activating chromosomal translocations. 1-4 In all 3 cases, the juxtaposition of c-myc to IgH (immunoglobulin heavy chain locus) was the most frequent event affecting >90% of the tumors, while translocations of c-myc to the Ig light chains (Igl or Igk) occurred at much lower frequencies (5%-10%). 5 Ig enhancers when translocated to c-myc drive expression of the c-myc gene in B cells, leading to tumor development.
Pristane-induced mouse plasmacytomas (PCTs) develop very slowly with a mean latency of 210 to 220 days. 6,7 Thus, c-Myc deregulation by chromosomal translocation (typically T(12;15)) 5 is an indispensable early initiating event in transformation but is not sufficient for the conversion of precursor cells into an overt malignant PCT. However, the additional events required for full tumor development in pristane-induced mouse plasmacytomagenesis remain elusive.
In contrast to pristane only–induced PCTs, pristane + v-abl–induced PCTs display a significant change in the ratio of the typical T(12;15) translocation versus the variant T(16;15) and T(6;15) translocations. The ratio of typical versus variant translocations in pristane-induced PCTs was 73:8 (10.9%). In contrast, this ratio changed to 51:34 (66.6%) in pristane + v-abl–induced PCTs. 5 Such a significant change in the translocation ratio suggests that a different effect on maturation of precursor B cells occurs during v-abl/myc–induced plasmacytoma development. 5,8 Indeed, v-abl targets pre-B cells undergoing light chain rearrangement and blocks their differentiation. 9-11
It is of note that when v-abl and myc genes are overexpressed in precursor pre-B cells, this combination no longer drives the development of lymphomas but rather induces PCTs. This is in contrast to v-abl overexpression alone that induces lymphomas. 8,12 This alternate pathway was identified in a series of transfer experiments, in which pre-B cells from Eµ-myc transgenic mice were infected with v-abl and transferred to compatible hosts, where they developed into plasmacytomas in the new host. 13,14 The same pre-B cells will exclusively generate lymphomas in these mice when v-abl is not coexpressed. 15,16 Similarly, cytogenetic and molecular analyses of spontaneous PCTs in v-abl transgenic mice revealed a rearranged, constitutively expressed c-myc gene due to spontaneous translocation to the Igh locus. 17 In these PCTs, both oncogenes were overexpressed.
Furthermore, our PCT induction studies using chimeras and radiochimeras of DBA2 newborn or sublethally irradiated adult DBA2 mice reconstituted with v-abl–infected spleen and bone marrow–derived cells of B/cRb6;15 origin provided strong evidence for an altered tumorigenic role for v-abl. 18-20 The PCTs that developed in this unusual induction system were of donor origin, and the majority were carriers of lambda/myc T(15;16) translocations. These translocations had not been observed using previous induction systems. In all of the above PCTs, both myc and v-abl were overexpressed.
The chimera experiments indicated that constitutive activation of the myc gene may have broadened the spectrum of B cell types that are susceptible to the abl viral infection. Myc also endowed on v-abl ability to transform precusor B cells into plasmacytomas instead of lymphomas. These studies gave the first evidence for a new tumorigenic function of v-abl and myc in PCT development in general and tumor acceleration in particular. 21
Pristane-induced PCTs that are followed by Abelson murine leukemia virus (A-MuLV) infection show shorter latencies and display overexpression of both c-myc and v-abl genes. These fast-onset PCTs develop with a mean latency of 36 days. 6,7 In addition to Ig/myc juxtaposition, these PCTs displayed a second nonrandom numerical chromosome aberration, trisomy of chromosome 11 (Chr 11). In pristane only–induced PCTs, trisomy 11 is found at a low frequency (7.1%), 22,23 whereas in pristane + v-abl–induced PCTs, the frequency of chromosome 11 trisomy increases to 61.1%. 24 The highest frequency of chromosome 11 changes in PCTs, up to 90%, 25 was found by pristane pretreatment of BALB/c or BALB/c congenic mice, infected with a helper-free v-abl/myc retrovirus. 7 In these experiments, the mean latency of PCT development was 45 days. 25 In these PCTs, the constitutive retroviral Myc expression suppresses the endogenous c-Myc production, and consequently, there is a lack of c-myc/Ig translocations. 26
Cytogenetic mapping of v-abl/myc–induced PCTs using F1 mice that were carriers of reciprocal translocations of chromosome 11, such as T(2;11)4Dn and T(11;16)53Dn, tentatively narrowed down the duplicated regions of chromosome 11 to band 11E. 25 Thus, the regular occurrence of chromosome 11 aberrations and particularly the duplication of band 11E led us to hypothesize that the nonrandom duplication of a portion of the 11E band contributes to an increase in gene dosage that is causally linked to the accelerated tumor development in v-abl/myc–induced PCTs.
The aim of the present study was to determine which subregion(s) of the telomeric 11E band is consistently duplicated in PCTs. To this end, we have used T38HxBALB/c mice, which carry reciprocal T(X;11) chromosomes with the telomeric 11E band translocated to the A2 band of chromosome X. Classic and molecular cytogenetic analyses (G-banding, chromosome painting, mBANDing) were carried out to establish that duplication of the 11E2 subcytoband of chromosome 11 was found in all PCTs.
Results
Characterization of the T38H mouse that carries a reciprocal translocation between chromosomes X and 11
The current study uses the T38H mice, 27,28 which carry a balanced translocation rcp(TX;11) that is illustrated in Figure 1. Figure 1A shows the location of the T38 breakpoints at band 11E on Chr 11 and at the A2 subband attached to the centromere of Chr X. The reciprocal translocation between these 2 chromosomes resulted in a long T(11;X) and a short T(X;11) chromosome. The long T(11;X) chromosome contains the centromere, the A, B, C, and D bands proximal to the T38H breakpoint, and the X-derived chromosomal segment distal to the A2 breakpoint. The short T(X;11) chromosome was generated by the translocation of the telomeric 11E band of Chr 11 onto the centromeric A2 subband of Chr X (distal to the A2 breakpoint).
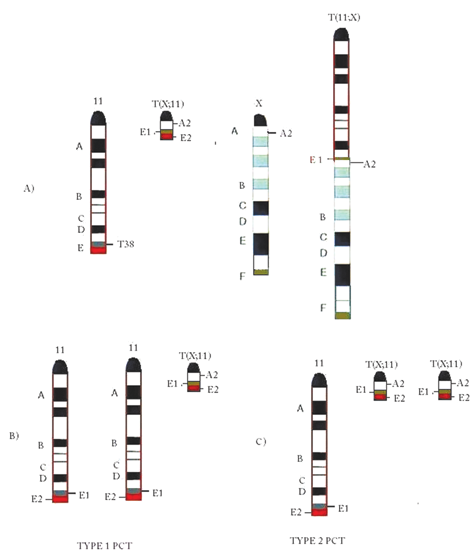
Graphical illustration of the chromosomal constitution of chromosomes 11 in the (BALB/cxT38H) F1 N backcross generation mouse and in type 1 and type 2 plasmacytomas (PCTs). (
Figure 1B and 1C illustrate the expected duplication patterns of chromosome 11 in v-abl/myc–induced PCTs. Based on the type of chromosome 11 duplication, we designate the expected PCTs as type 1 or type 2. In type 1 PCT (Fig. 1B), the trisomy of the specific segment of Chr 11 will consist of 2 copies from the normal diploid chromosome Chr 11 of BALB/c origin and the third copy from the end segment of the short T(X;11) chromosome. In the type 2 PCT (Fig. 1C), the trisomy of the 11E band results from the duplication of the T(X;11) chromosome and a third copy of the normal intact Chr 11 of BALB/c origin.
The prediction of the 2 types of PCT tumors was based on the position of the translocation breakpoints on the 11 and X chromosomes. Thus, the possibility of trisomy due to the duplication of the T(11;X) chromosome was excluded a priori due to previous cytogenetic mapping studies of PCTs in mice carrying reciprocally translocated chromosomes T(2;11) and T(2;16). 25 These previous results strongly suggested that the duplication of segments of Chr 11 distal to band D were needed for accelerated PCTs, 25 therefore eliminating the T(11;X) chromosome as a possible duplication target giving rise to trisomy 11.
Cytogenetic analysis of v-abl/myc–induced PCTs
The chromosome constitution of a normal female (BALB/cxT38H) N backcrossed mouse that was used for v-abl/myc– induced tumors is shown in Figure 2A. Figure 2B shows the G-banding results that identify the T(X;11) and T(11;X) chromosomes as well as the normal X and 11 chromosomes of BALB/c origin. Chromosome 11 painting confirmed the presence of Chr 11 material on the translocated chromosomes T(11;X), T(X;11), and the normal Chr 11 (Fig. 2C).
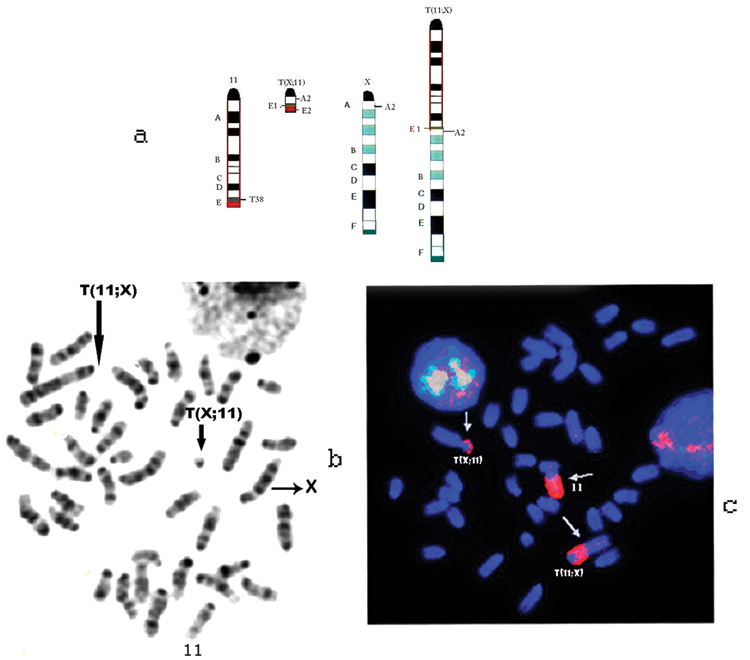
G-banded and chromosome 11–painted metaphase plates of female BALB/cxT38H N mice used in this study. (
Thirty-five v-abl/myc–induced primary and first generation transplanted PCTs were analyzed with respect to tumor latency and chromosomal 11 constitution by FISH and occasionally by G-banding (Table 1 and Suppl. Table S1). Thirteen tumors were also examined by mBANDing. These PCT results are summarized in Table 1. The PCT type was based on the analysis of 20 metaphases that displayed identical chromosome 11 paint hybridization patterns. We found either trisomy of chromosome 11 (type 1) or the duplication of the T(X;11) chromosome (type 2) in the PCTs; type 2 PCTs were more frequent than type 1 PCTs (Suppl. Table S1). The duplication pattern of the T(X;11) chromosome induced in 2 males with a XXY sex chromosome constitution (Klinefelter) did not differ from those found in females. However, since few surviving male mice had any sex chromosome aberrations, they were excluded from further tumor induction experiments.
Characterization of v-abl/myc–Induced Plasmacytomas
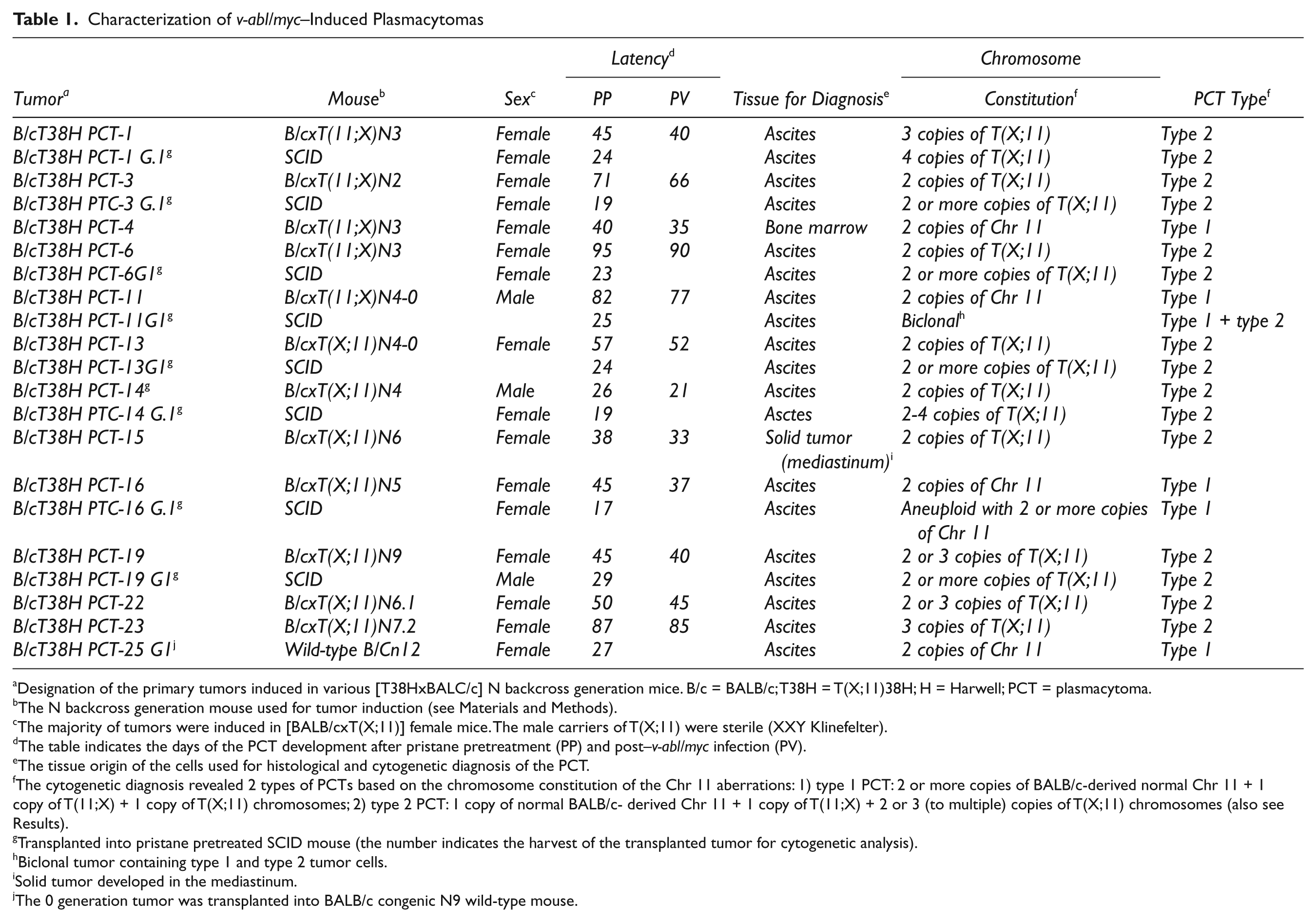
Designation of the primary tumors induced in various [T38HxBALC/c] N backcross generation mice. B/c = BALB/c; T38H = T(X;11)38H; H = Harwell; PCT = plasmacytoma.
The N backcross generation mouse used for tumor induction (see Materials and Methods).
The majority of tumors were induced in [BALB/cxT(X;11)] female mice. The male carriers of T(X;11) were sterile (XXY Klinefelter).
The table indicates the days of the PCT development after pristane pretreatment (PP) and post–v-abl/myc infection (PV).
The tissue origin of the cells used for histological and cytogenetic diagnosis of the PCT.
The cytogenetic diagnosis revealed 2 types of PCTs based on the chromosome constitution of the Chr 11 aberrations: 1) type 1 PCT: 2 or more copies of BALB/c-derived normal Chr 11 + 1 copy of T(11;X) + 1 copy of T(X;11) chromosomes; 2) type 2 PCT: 1 copy of normal BALB/c- derived Chr 11 + 1 copy of T(11;X) + 2 or 3 (to multiple) copies of T(X;11) chromosomes (also see Results).
Transplanted into pristane pretreated SCID mouse (the number indicates the harvest of the transplanted tumor for cytogenetic analysis).
Biclonal tumor containing type 1 and type 2 tumor cells.
Solid tumor developed in the mediastinum.
The 0 generation tumor was transplanted into BALB/c congenic N9 wild-type mouse.
Examples of the chromosome constitution of type 1 and type 2 tumors as determined by G-banding and FISH staining are shown in Figures 3 and 4, respectively. Type 1 PCTs are based on the presence of 2 copies of chromosome 11 and 1 copy of the T(X;11) chromosome (Fig. 3A and B), whereas type 2 PCTs show the presence of 2 copies of the T(X;11) chromosome and 1 copy of the normal chromosome 11 (Fig. 4A and B). It should be noted that the PCT types refer to the cytogenetic mechanism leading to trisomy of the 11E band. Irrespective of these PCT types, all pristane + v-abl/myc–induced PCTs showed trisomy of Chr 11E.
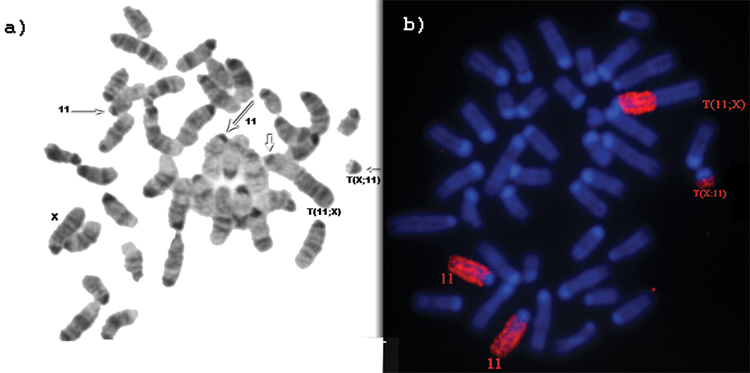
G-banded and chromosome 11–painted metaphase plates of type 1 PCT. (
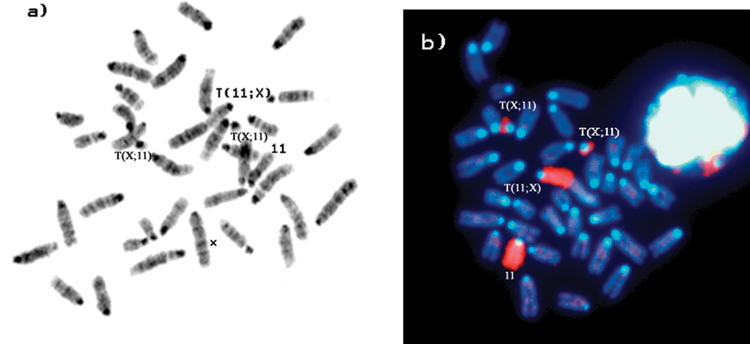
G-banded and chromosome 11–painted metaphase plates of type 2 PCT. (
The duplication pattern of type 1 and type 2 tumors revealed that trisomy of the 11E band was always present in all analyzed tumors (Table 1). However, in both types of PCTs and especially in those of the first transplant generation, there were more than 3 copies of the 11E band present in a number of metaphase plates. Figure 5B shows a metaphase plate in which both the T(X;11) and the BALB/c-derived chromosome 11 are duplicated, providing the cell with 4 copies of the 11E band, likely of type 1 origin. Similarly, in Figure 5A, chromosome 11 painting revealed 3 copies of T(X;11) chromosomes plus a normal chromosome 11, resulting in a cell with 4 copies of the 11E band, suggesting a type 2 origin. As expected, amplification of the T(11;X) chromosome was not identified in any of the tumors analyzed. In contrast, the G-banding and chromosome 11 painting uncovered some rare cells missing the T(11;X) chromosome, consistent with the conclusion that chromosome 11 segments proximal to the T38H breakpoint are not necessary for accelerated PCT development (Fig. 5C). In a few polyploid tumor cells, the amplification rate of the T(X;11) chromosome was maintained between 3 to 4 copies per diploid constitution of the polyploid cell. Figure 5D shows multiple copies of the small T(X;11) chromosome. Biclonality, that is, PCTs of types 1 and 2 within the same tumor sample, was occasionally observed (Table 1 and Suppl. Table S1).
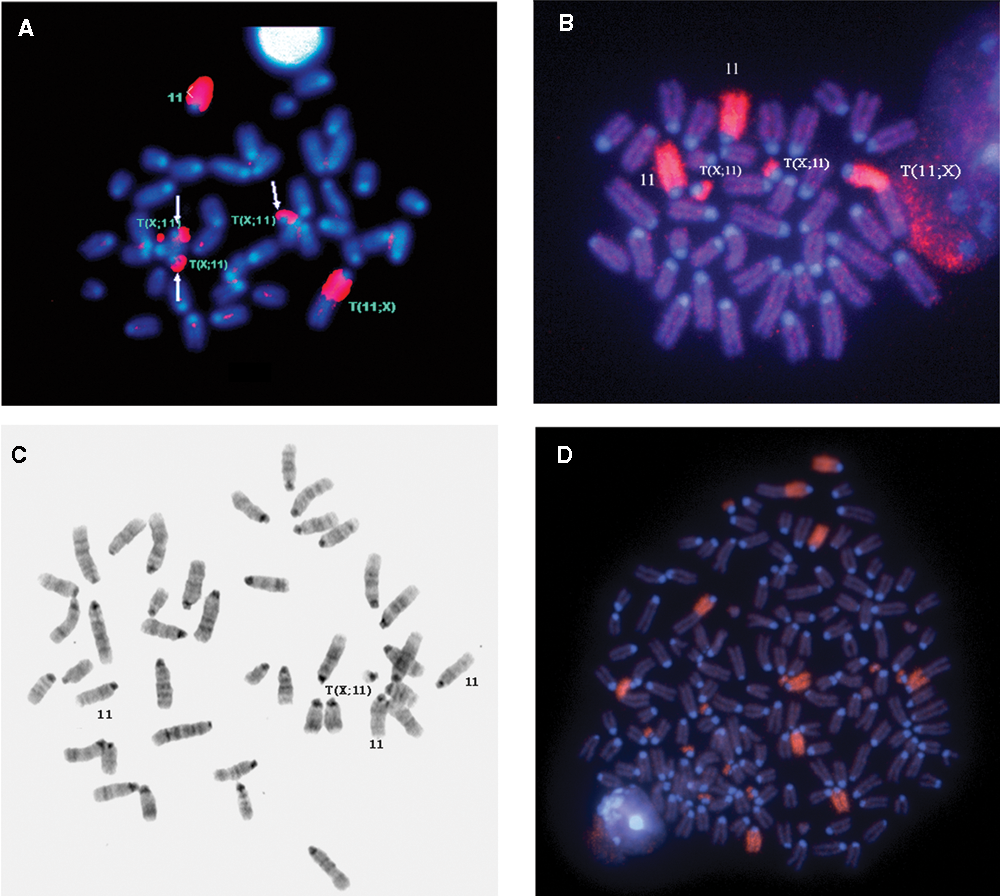
Metaphase plates with multiple telomeric 11E bands. (
mBANDing confirmed cytoband 11E2 as the critical trisomic segment always associated with the v-abl/myc tumorigenesis
In cytogenetic terms, the 11E band distal to the T38H breakpoint is a complex chromosomal unit. It contains a part of the 11E1 subband distal to the T38 breakpoint and the whole 11E2 subband from Chr 11. This chromosomal region is attached to the segment of the A2 band of Chr X located distal to the A2 breakpoint. To better define the chromosomal organization of the 11E subband on the T(11;X) and T(X;11) chromosomes, we used multicolor banding (mBANDing), which allows for the analysis of intrachromosomal rearrangements. 29 We had previously developed mBANDing for mouse chromosome 11 and especially focused on the telomeric 11E band. 36
Using mBAND11 probes for chromosome 11, we examined the nature of the chromosome 11 duplication. Figure 6A illustrates the mBAND labeling scheme for mouse chromosome 11. Figure 6B shows the mBAND profile for the normal (T38HxBALB/c) N mouse. In this mouse, the 11E band is present as a single copy segment in the end bands of the normal chromosome 11 and as a smaller band, due to the breakpoint in E1, 28 in both the T(X;11) and the T(11;X) chromosomes. Figure 6C and 6D highlight the mBAND patterns of the 2 types of duplications observed in primary (generation 0) v-abl/myc–induced PCTs. These duplications involve either the whole chromosome 11, which is then present in 2 copies, or the small T(X;11) chromosome, which is found in 2 copies (Fig. 6D and 6C, respectively). In addition, we observed an interstitial (intrachromosomal) duplication of 11E2 (Fig. 6E, arrows). This alternate mechanism of 11E2 duplication was seen in 5 of 20 metaphases, that is, in 20% of the metaphases. The intrachromosomal duplication of 11E2 was more commonly found associated with the T(X;11) chromosome (4/20 metphases) than with the normal chromosome 11 (1/20 metaphases); however, it occurred in 11E2 of both chromosomes (Fig. 6E, arrows). Additionally, the usefulness of the mBANDing was underscored by its capability to reveal the presence of the interstitially duplicated 11E2 band also found in diploid metaphase plates, which were first considered of host cell origin based on G-banding and chromosome painting.
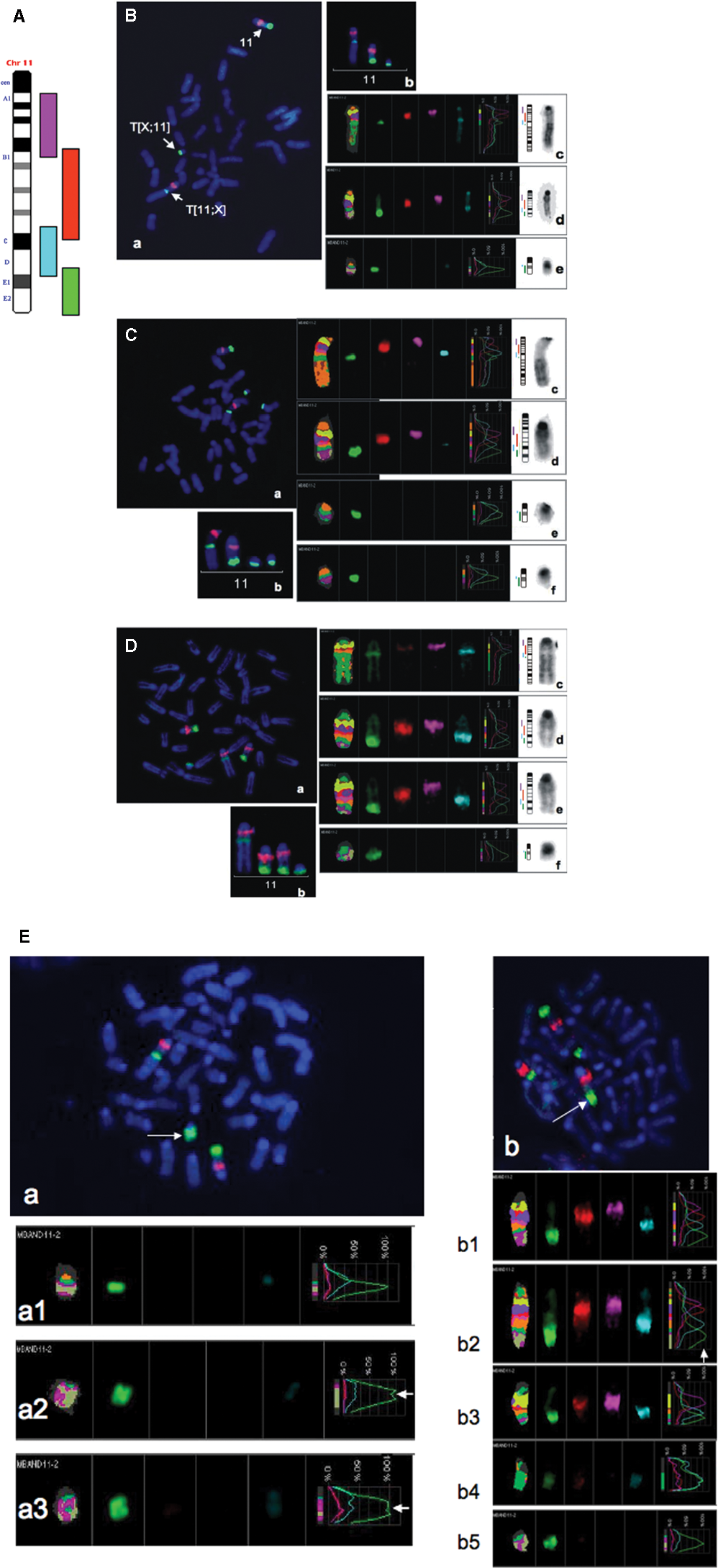
mBANDing of chromosome 11 in v-abl/myc–induced PCTs. (
Pristane only–induced tumors did not develop duplications of chromosome 11 or of 11E bands (Suppl. Table S2). In 6 pristane only–induced PCTs, chromosome 11 was unchanged, with one exception: PCT 4078, which had in 4 of 21 metaphases an insertion of 11E2 into a new chromosomal location (Suppl. Table S2). However, all chromosomes 11 in this PCT were normal based on mBANDing. These results further support the importance of subregion 11E in accelerated PCT development.
Thus, the cytogenetic pattern of the E2 duplication is found as 3 forms: 1) the duplication of T(X;11) chromosome, 2) the duplication of chromosome 11, and 3) the intrachromosomal duplication of the 11E2 band on either Chr 11 or T(X;11). A duplication of T(11;X) was never seen. We conclude from these results that 11E2 is the chromosomal segment that contributes to accelerated PCT development in pristane + v-abl/myc–induced PCTs.
Discussion
11E2 subband is always trisomic in v-abl/myc PCT genesis
The present study focused on determining which segment of chromosome 11 is critical for the accelerated development of PCTs induced by the v-abl/myc oncogenes. Using classic and molecular cytogenetics, we ascertained that the 11E2 subcytoband was the critical segment on chromosome 11. This region was always found duplicated in the v-abl/myc–induced plasmacytomas. In all 13 v-abl/myc–induced PCTs, the presence of at least 3 copies of the 11E2 subband was found in each analyzed metaphase plate, irrespective of whether they were type 1 or type 2 PCTs.
Previous findings had shown that trisomy of Chr 11 was present in more than 95% of the rapidly developing plasmacytomas induced in pristane-primed BALB/c mice with Ig/myc translocations and Abelson leukemia virus infection (ABPC tumors). 24 One suggestion was that the shortened latency might be attributed merely to the interaction of activated c-myc and v-abl oncogenes. It was hypothesized that this oncogene interaction disrupts cell cycle controls, thus bypassing the multiple genetic changes that are normally required for the long-term pristane transformation process (TEPC tumors). 5,24 On the other hand, the finding of the nonrandom duplication of Chr 11 segments distal to band D of Chr 11 in v-abl/myc PCTs argued strongly for a possible causal effect of this numerical chromosomal aberration in accelerated PCT genesis. 25
Moreover, previous cytogenetic investigations revealed that v-abl–induced lymphoid tumors were diploid or near-diploid without any nonrandom structural and/or numerical chromosomal aberrations. 30,31 The only exceptions were lymphomas with trisomy of chromosome 15. 32-35 This implies that the v-abl oncogene on its own is insufficient to cause selection for chromosome 11 trisomy. Similarly, c-Myc alone induces a very low frequency of Chr 11 aberrations (7.1%). 22,23 Overexpression of both v-abl and myc drives accelerated plasmacytoma development, 5 and these tumors have the highest incidence of chromosome 11 aberrations. 25 Thus, it is the combined effect of both genes that selects for chromosome 11 changes to allow for accelerated plasmacytomas. These chromosomal changes on chromosome 11 have now been attributed to the duplication of 11E2.
The duplication of cytoband 11E (and subband E2) can occur by several mechanisms, such as duplication of the whole chromosome 11 or a part of it, such as a centromeric-fused isochromosome 11. Other mechanisms include translocation to another chromosome or as an extrachromosomal element. 36 In the present study, the prevailing forms found were the duplication of the whole of chromosome 11 or a segment of it as part of the T(X;11) chromosome. Tumor cells with increased copy number of the 11E band were frequently revealed by the cytogenetic analyses (Figs. 3-6). One can conclude that the amplification of this band is a consistent finding in the v-abl/myc–induced tumors and thus contains the region involved in v-abl/myc induction of accelerated PCT development.
mBANDing studies allowed us to define more accurately the trisomic status of the 11E band. It was found that the 11E2 subband was always present as a trisomic entity or greater in v-abl/myc–induced PCTs. The main findings that validate this finding were that 1) in pristane alone–induced primary or polyploid secondary tumors, the copy number of the 11E band was 2 or multiple of 2 (Suppl. Table S2); and 2) in some diploid or near-diploid metaphase plates of v-abl/myc–induced PCTs with only 2 chromosome 11s, generally considered as normal cells infiltrating the tumor, some cells had interstitial duplication of the 11E band (Fig. 6). This indicates that these were true tumor cells.
The synteny data argue indirectly for the oncogenic and accelerating function of the genes located on cytoband 11E2
Synteny data revealed that the cytoband 11E2 is present in specific chromosomes that are involved in the progression of other tumors of human and rodent origin. The human synteny region for mouse Chr 11E2 is on chromosome 17q25, and the rat synteny region is on 10q32. Region 17q25 is commonly involved in duplication, amplification, or translocation events in human tumors. In human acute myeloid leukemia M4, region 17q25 is involved in translocations, t(13;17)(q14;q25), which are associated with poor prognosis. 37 A fusion gene in acute myeloid leukemia (AML) also involves the chromosomal region in question (17q25), such that AF17q25 is fused with the MLL gene in AML with t(11;17)(q23;q25) translocations. 38 In chronic myeloid leukemia, breakpoint clusters have been reported around 17q25. 39 17q25 amplifications are common in breast, ovarian, and thyroid cancers, neuroblastomas, and osteosarcoma. 40-47 The rat 10q32 region was shown to be involved in schwannomas. 48
Although the critical genes amplified in the 11E2 cytoband have not been identified to date, further experiments such as array comparative genome hybridization and functional genetic analyses may aid in their identification. These experiments are now in progress. In summary, mouse chromosome 11 band E2 and the synteny regions in rats and humans seem to be critical players in the tumorigenic process. Illuminating their newly acquired function due to duplication will aid our understanding of the significance of this phenomenon that frequently occurs in many tumors of different types.
Materials and Methods
Mouse strain
The mouse strain used in the present studies was the T38H induced by acute X irradiation (1000-1200 rad) of spermatozoa of F1 (C3H female x 101 male) 27,28 and kept as frozen embryos at MRC-Harwell (Oxfordshire, UK). 49 We obtained 25 T38H embryos with 4 possible phenotypes resulting from a cross T38H +/+ Otcspf (sparse fur)X with OtcspfY. For our purposes, only the hemizygous T38H +/+ spf wild-type females were useful since they were fertile and were carriers of the reciprocal translocation between chromosomes 11 and X. The wild-type T38H +/+ Y males had reduced fertility or were completely sterile; therefore, the strain could only be maintained as hemizygous T38H females. 27,28,49
The embryos were rederived at Jackson Labs (Bar Harbor, Maine, USA), and the rederivation resulted in 2 (T38H × OTCspf) females and 1 sterile (XXY) male. A breeding colony was set up with 2 female carriers of the T(X;11) reciprocally translocated chromosomes mated with BALB/c males. This was done in order to expand the number of heterozygote female carriers of the reciprocal translocation and to obtain N congenic heterozygote generation female mice with higher BALB/c background, increasing their susceptibility for plasmacytoma genesis (protocol #007-002 approved by Central Animal Care).
Retrovirus
A highly plasmacytomagenic retroviral construct (obtained from Neoclone, Madison, WI), which expresses c-myc and v-abl oncogenes, was used for the infection of pristane-conditioned N congenic mice. The construction and production of the v-abl/myc retrovirus were described previously. 7
Tumor induction, diagnosis, and tumor transplantation
A total number of 1,013 offspring (363 males and 650 females) were obtained from N1 to N14 generation backcrosses. The male mice were either wild-type or carriers of the reciprocal translocation but had acquired an additional X chromosome, thus creating males with a Klinefelter-type genome. Therefore, males, with a few exceptions, were discarded.
All females were first karyotyped by Giemsa staining (Karyomax Giemsa Stain, Gibco/Invitrogen, Carlsbad, CA) for the presence of the reciprocally translocated T(X;11) chromosomes in metaphases of peritoneal derived cells. The mice with plates containing the small T(X;11) marker chromosome (easily identifiable due to its minute size) were further analyzed by G-banding and/or FISH staining to confirm that they were carriers of the rcp(X;11) chromosomes, thus suitable for tumor induction (see Results). Only about 25% of all females born were found to be carriers of the reciprocally translocated chromosome T(X;11).
Thirty-five adult (T38HxB/c) N2 to N11 mice (8 weeks of age) were pretreated with a single intraperitoneal (IP) injection of 0.3 mL of pristane. Five days after pristane treatment, the mice were inoculated IP with 0.5 to 1 mL of v-abl/myc virus (104 ffu). The primary tumors that developed after a short latency were of different types but mostly as ascitic tumors in the peritoneal cavity or as solid tumors often localized both in the peritoneal cavity and in the mediastinum (Table 1 and Suppl. Table S1). The diagnosis of PCTs was based on morphological criteria using a modified Giemsa stain. For transplantation experiments, 1 × 106 cells were IP injected into pristane-pretreated SCID mice (8 weeks old). The tumor-derived cells were subjected to classic and molecular cytogenetic analysis.
Classic and molecular cytogenetics
Metaphase spreads were prepared from ascitic plasmacytomas, oil granuloma, or solid tumor cells without colcemid treatment. 50 Twenty metaphases were examined per mouse. Chromosome identification of the G-banded plates followed the recommendations of the Committee of Standard Genetic Nomenclature for Mice Mouse News Letter. 51
Chromosome painting
Chromosome painting for chromosomes X and 11 and the translocated Chr 11 segments was performed as described 50 ; the paints were purchased from Applied Spectral Imaging Inc. (Vista, CA).
mBANDing
Mouse mBANDing for chromosome 11 was developed by our group previously. 36 Image acquisition was performed using 63x oil (1.4 aperture) objective (Carl Zeiss Ltd., Toronto, ON, Canada) and the ISIS-FISH imaging system 5.0 SR 3 (Metasystems Group Inc., Boston, MA) on an Axioplan 2 microscope (Carl Zeiss Ltd.). Chromosomes were prepared as previously published, 50 and mBANDing was performed as recommended by the supplier. Briefly, slides were treated with pepsin (50 µg/mL) in 0.01 M HCl for 2 minutes, washed in 1x PBS for 3 minutes, and placed in 1% formaldehyde in 1x PBS/MgCl2 for 10 minutes, after which they were washed in 1x PBS for 3 minutes. Denaturation of the slides was carried out in 0.1x SSC at room temperature for 1 minute. The slides were then incubated in 2x SSC at 70°C for 30 minutes. The coplin jar containing the slides was removed from the water bath and cooled down to 37°C. The slides were transferred into 0.1x SSC at room temperature for 1 minute, denatured in 0.07 M NaOH at room temperature for 1 minute, placed into 0.1x SSC at 4°C for 1 minute, then into 2x SSC at 4°C for 1 minute, dehydrated by increasing the percentages of ethanol for 1 minute each (30%, 50%, 70%, and 100%, respectively), and air dried. The mBAND probe was denatured at 75°C for 5 minutes, placed on ice briefly, incubated at 37°C for 30 minutes, and then hybridized onto the metaphase plates at 37°C for 2 days. The posthybridization washes included 1x SSC at 75°C for 5 minutes, followed by incubation of the slides in 4x SSC with 0.05% Tween 20 at room temperature and a wash in the same buffer for 3 minutes. The slides were then drained and counterstained with DAPI (1 µg/mL) and mounted in antifade (Metasystems Group Inc.).
Footnotes
Acknowledgements
The authors thank Dr. George Klein for ongoing discussions and interest, MRC-Harwell (Oxfordshire, UK), and especially Peter Glenister for the T38H frozen embryos and his advice on the recovery of the embryos.
This work was funded by a grant from the Canadian Institutes of Health Research to SM grant #89755, by the Cancer Research Society (MRAM) and CancerCare Manitoba Foundation (MRAM).
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.