
Abstract
The ataxia-telangiectasia mutated (ATM) kinase is activated in the cellular response to ionizing radiation (IR) and is of importance to the repair of DNA double-strand breaks (DSBs). The MUC1 oncoprotein is aberrantly overexpressed in human breast carcinomas. The present work demonstrates that the MUC1 C-terminal subunit (MUC1-C) constitutively interacts with ATM in human breast cancer cells. The authors show that the MUC1-C cytoplasmic domain binds directly to ATM HEAT repeats. The results also demonstrate that the MUC1-C cytoplasmic domain binds to the ATM substrate H2AX. The functional significance of these interactions is supported by the finding that MUC1-C promotes removal of IR-induced nuclear γH2AX foci. MUC1-C also protects against IR-induced chromosomal aberrations. In concert with these results, MUC1-C blocks IR-induced death by promoting repair of potentially lethal DNA damage. These findings indicate that the overexpression of MUC1 can protect against IR-induced DNA DSBs and may represent a physiologic response that has been exploited by malignant cells.
Introduction
The ataxia telangiectasia mutated (ATM) kinase is activated in the cellular response to ionizing radiation (IR). Mutation of ATM is associated with genomic instability, increased sensitivity to IR, and cell cycle defects in the response to DNA damage. 1 ATM phosphorylates diverse targets, such as H2AX, NBS1, and Chk1, that activate cell cycle checkpoints and DNA repair. 2 The phosphorylation of H2AX (γH2AX) is an early ATM-mediated response to DNA double-strand breaks (DSBs). 3-5 In turn, γH2AX interacts with NBS1 to form nuclear foci at sites of DNA damage. 6 The interaction between γH2AX and NBS1 then further contributes to recruitment of MRE11 and RAD50 from the cytoplasm to DNA DSB sites. 6,7 In this regard, loss of H2AX is associated with defects in DSB repair and genomic instability due to decreased formation of nuclear foci. 8,9 Activation of ATM and its recruitment to DNA DSBs also involves interaction with NBS1. 10 The HEAT repeats in ATM bind directly to the NBS1 C-terminal FXF/Y domain, and this interaction is essential for ATM activation. 10 In turn, the MRE11-RAD50-NBS1 (MRN) complex functions in ATM-mediated checkpoint responses and cell survival. 11-14 Early phosphorylation of H2AX is of importance to the formation of the MRN complex and to induction of cell cycle arrest and DNA repair. 3 The kinetics of H2AX phosphorylation also reflect the rate of DSB repair and resolution of the DNA damage response. 3,15
Mucin 1 (MUC1) is a heterodimeric transmembrane protein that is aberrantly overexpressed in diverse human carcinomas. 16,17 The MUC1 C-terminal subunit (MUC1-C) consists of a 58–amino acid extracellular domain, a 28–amino acid transmembrane domain, and a 72–amino acid cytoplasmic tail. 17,18 Galectin-3 interacts with the MUC1-C extracellular domain and promotes the formation of MUC1-C complexes with growth factor receptors. 19 The MUC1-C cytoplasmic domain is phosphorylated by c-Src, 20 protein kinase C, 21 glycogen synthase kinase 3β, 22 and c-Abl. 23 Moreover, the MUC1-C cytoplasmic domain interacts with effectors, such as β-catenin, 24,25 IκB kinase, 26 and p53, 27 which have been linked to the regulation of cell growth and death. Overexpression of MUC1 results in the accumulation of MUC1-C in the cytoplasm and targeting of this subunit to the nucleus 27-30 and mitochondria. 31,32 Importantly, expression of the MUC1-C cytoplasmic domain is sufficient to induce transformation 24 and activate expression of gene signatures associated with poor outcomes in patients with breast cancer. 33,34 Other studies have demonstrated that MUC1-C confers resistance to apoptosis or necrosis in the response of cells to (1) DNA damage induced by chemotherapeutic agents, 31,35 (2) reactive oxygen species (ROS)–induced stress, 36,37 and (3) hypoxia. 38 IR activates diverse forms of ROS, among which hydroxyl radicals contribute to the formation of DSBs. However, it is not known if MUC1 affects the response to IR treatment.
The present results demonstrate that MUC1-C associates with ATM and that the MUC1-C cytoplasmic domain binds directly to the ATM HEAT repeat region. The results also demonstrate that MUC1-C interacts with H2AX, promotes removal of IR-induced γH2AX foci, and protects against IR-induced cell death.
Results
MUC1-C associates with ATM
To investigate the potential interaction between MUC1 and the DNA damage response, we first studied the interaction between MUC1-C and ATM in human ZR-75-1 breast cancer cells that overexpress endogenous MUC1. Immunoblot analysis of anti-MUC1-C precipitates with anti-ATM demonstrated that MUC1-C associates with ATM (Fig. 1A, left). These results were confirmed in the reciprocal experiment by immunoblotting anti-ATM precipitates with anti-MUC1-C (Fig. 1A, right). The coprecipitation of MUC1-C and ATM was also obtained in studies of MCF-7 breast cancer cells (Fig. 1B). In addition, transient transfection of 293 cells with a Flag-tagged ATM and MUC1 confirmed that MUC1-C associates with ATM in reciprocal immunoprecipitation experiments (Fig. 1C). To determine whether the MUC1-C cytoplasmic domain (MUC1-CD) confers the association, we expressed green fluorescent protein (GFP)–tagged MUC1-CD in 293 cells. Immunoblot analysis of anti-ATM precipitates with anti-GFP further demonstrated that MUC1-CD forms complexes with ATM (Fig. 1D). These findings indicated that MUC1-C associates with ATM and that the interaction is conferred through the MUC1-C cytoplasmic domain.
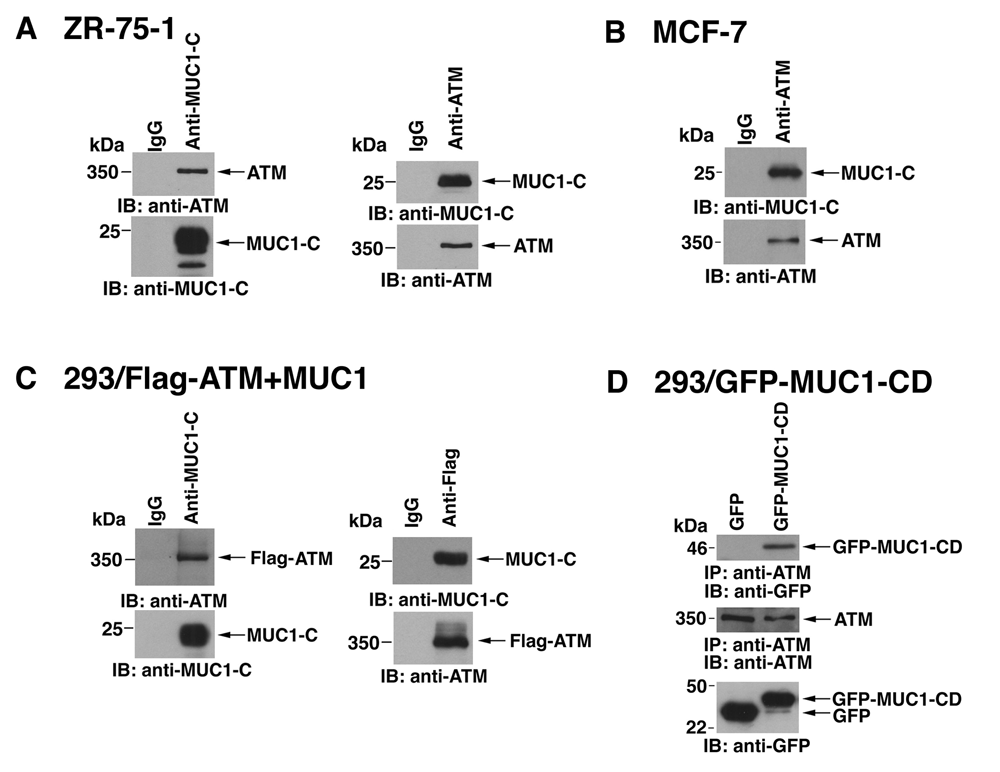
MUC1-C associates with ataxia-telangiectasia mutated (ATM) kinase. (
MUC1-CD binds directly to ATM
To define the region of ATM responsible for the interaction with MUC1-C, we purified glutathione S-transferase (GST) fusion proteins containing fragments of ATM (Fig. 2A). 39 Incubation of the GST and the GST-ATM proteins with lysates from 293 cells expressing GFP-MUC1-CD demonstrated binding of MUC1-CD to ATM(1439-1770), ATM(1764-2138), ATM (2141-2428), and ATM(2842-3056) (Fig. 2B). To determine whether the interaction is direct, we incubated the GST-ATM proteins with purified His-tagged MUC1-CD. The results demonstrate that MUC1-CD binds directly to ATM(1439-1770) and not the other fragments (Fig. 2C). Experiments with deletion mutants of MUC1-CD further demonstrated that the N-terminal region (amino acids 1-5; CQCRR) is sufficient for binding to ATM(1439-1770) (Fig. 2D). These findings demonstrate that MUC1-CD binds directly with the HEAT repeat region of ATM.
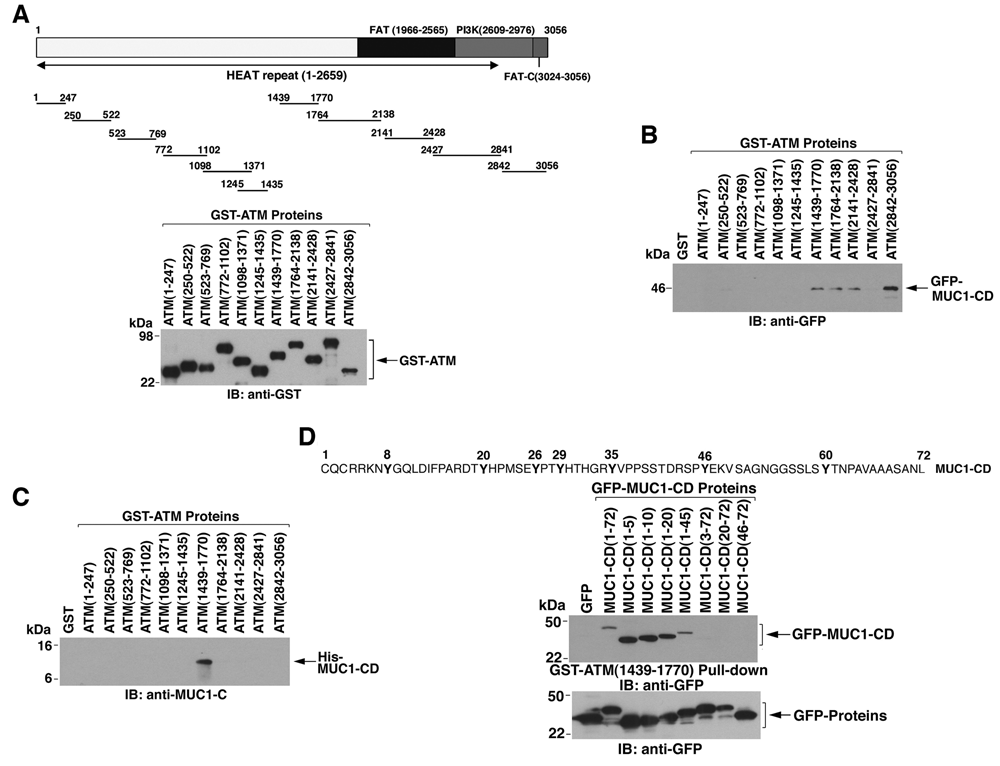
MUC1-CD binds directly to ataxia-telangiectasia mutated (ATM) kinase. (
MUC1-C CQC motif is responsible for interactions with ATM and H2AX
To more precisely define the MUC1-CD amino acids responsible for the interaction, we mutated Cys-1 to Ala (C1A) and Cys-3 to Ala (C3A). Binding of the MUC1-CD(C1A) mutant to ATM(1439-1770) was somewhat increased compared to that obtained with MUC1-CD (Fig. 3A). Moreover, the association with ATM(1439-1770) was decreased with MUC1-CD(C3A) and abrogated with the double MUC1-CD(C1A/C3A) mutant (Fig. 3A). Mutation of both Cys-1 and Cys-3 also abrogated the association of MUC1-CD with ATM(1764-2138), ATM(2141-2428), and ATM(2842-3056) (Fig. 3B). These results indicated that MUC1-CD amino acids Cys-1/Cys-3 confer direct binding to ATM(1439-1770) and that MUC1-CD associates indirectly with ATM(1764-2138), ATM(2141-2428), and ATM(2842-3056) in cells. ATM phosphorylates H2AX on Ser-139 in the response to DNA damage. 3,40 To determine whether MUC1-C also associates with H2AX, lysates from 293 cells expressing GFP or GFP-MUC1-CD were incubated with GST-H2AX. Immunoblot analysis of the adsorbates demonstrated that H2AX associates with MUC1-CD (Fig. 3C). In addition, incubation of GST-H2AX with lysates of 293 cells expressing GFP-MUC1-CD mutants demonstrated that H2AX interacts with MUC1-CD(1-45) but not MUC1-CD(46-72) or MUC1-CD(C1A/C3A) (Fig. 3C). Incubation of GST-H2AX with His-MUC1-CD further demonstrated a direct interaction between MUC1-CD and H2AX (Fig. 3D). These findings indicate that MUC1-C associates with ATM and H2AX.
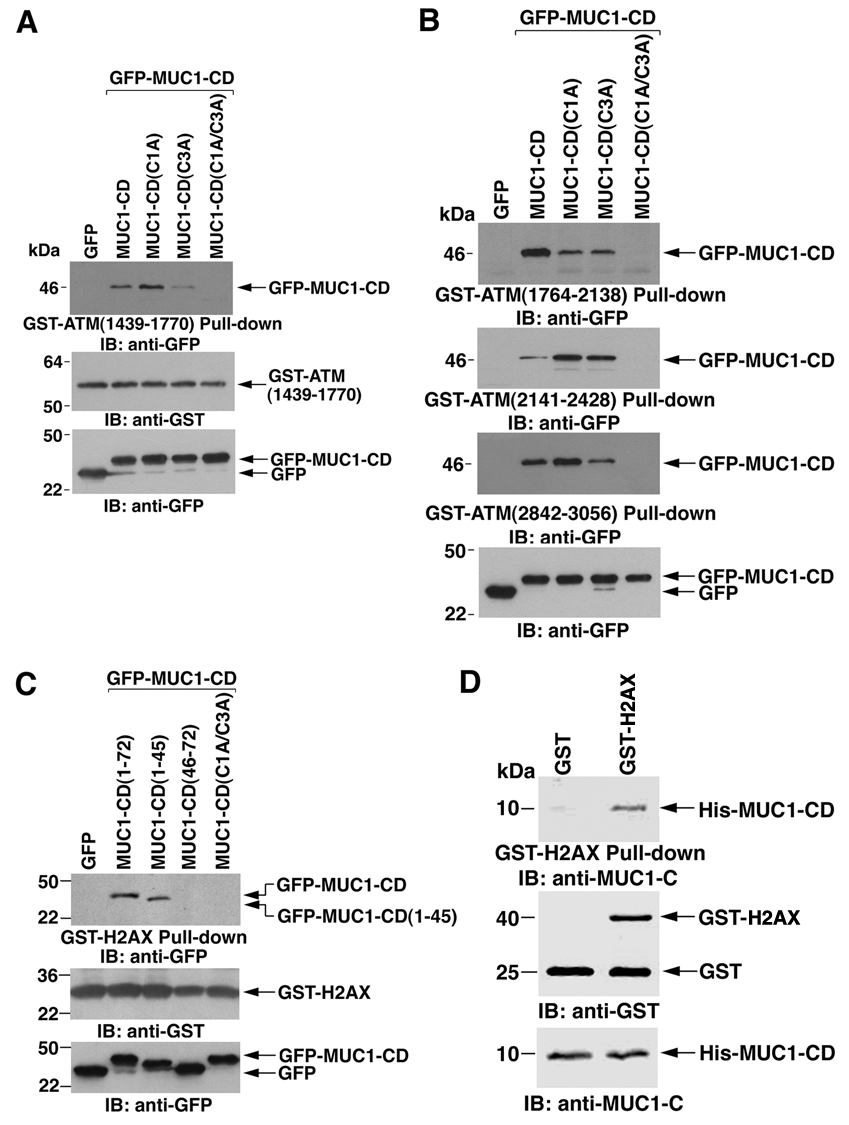
MUC1-C associates with the ataxia-telangiectasia mutated (ATM)–H2AX complex. (
MUC1 regulates ATM-mediated phosphorylation of H2AX
To assess whether MUC1 affects H2AX phosphorylation, we irradiated ZR-75-1 cells stably expressing an empty vector or a MUC1siRNA. 27,30,31 Notably, analysis of lysates from 2 ZR-75-1/MUC1siRNA cell clones exposed to 2.5 and 5.0 Gy IR demonstrated that silencing MUC1 is associated with increases in intensity of H2AX phosphorylation (Fig. 4A). ATM-mediated phosphorylation of H2AX is associated with activation of cell cycle checkpoints. 2,40,41 To determine whether MUC1 also affects checkpoint activation, we studied cell cycle distribution in the response to IR treatment. Silencing MUC1 in ZR-75-1 cells was associated with a marked increase in accumulation of S phase cells as compared to that in ZR-75-1/vector cells expressing endogenous MUC1 (Fig. 4B). In concert with these results, silencing MUC1 in ZR-75-1 cells increased the IR-induced arrest in G2-M phase as determined by phospho-histone H3 staining (Fig. 4C). To extend the analysis of MUC1 in the IR response, the ZR-75-1/MUC1siRNA cells were transfected with an empty vector or one expressing Flag-MUC1-CD (Fig. 4D). MUC1-CD suppressed the induction of γH2AX in response to 2.5 and 10 Gy IR (Fig. 4D). By contrast, this suppression of IR-induced γH2AX was attenuated by expression of the MUC1-CD(C1A/C3A) mutant that fails to associate with ATM and H2AX (Fig. 4D). These findings provided support for involvement of MUC1 in the regulation of IR-induced H2AX phosphorylation.
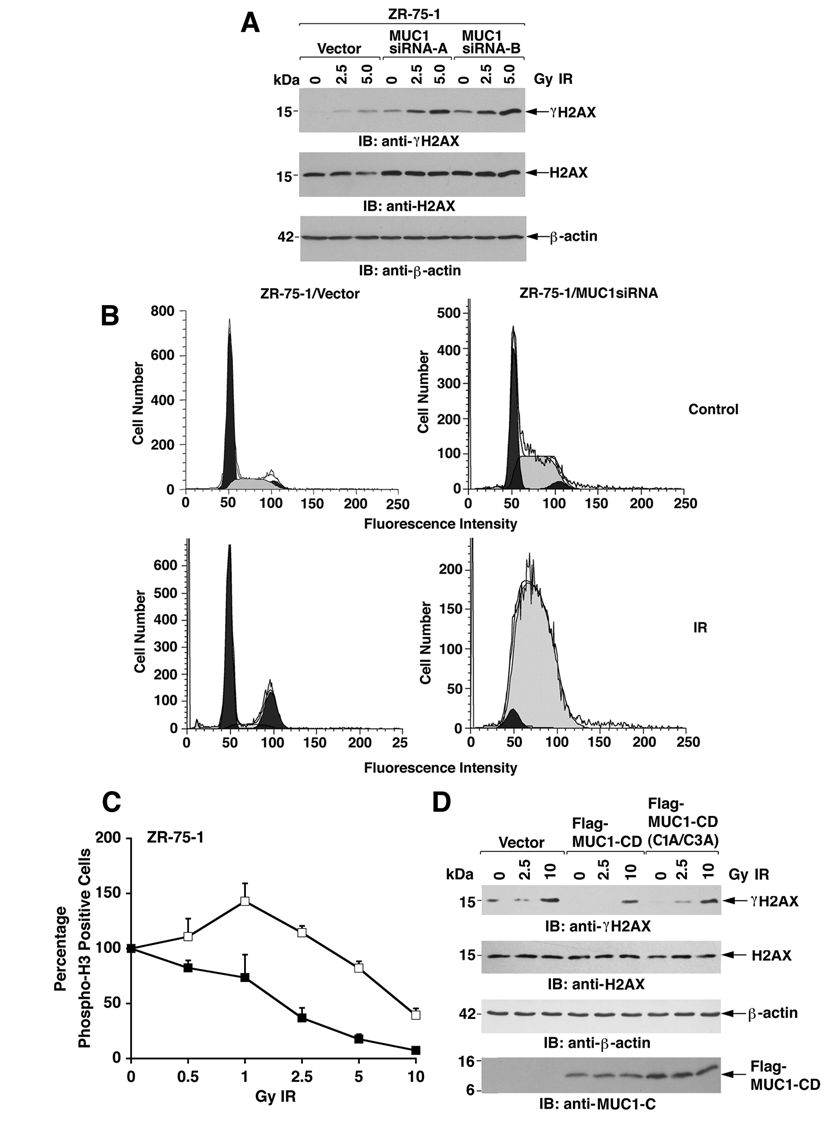
Silencing MUC1 is associated with increased levels of γH2AX in the response to ionizing radiation (IR). (
MUC1 promotes removal of γH2AX foci and attenuates loss of clonogenic survival
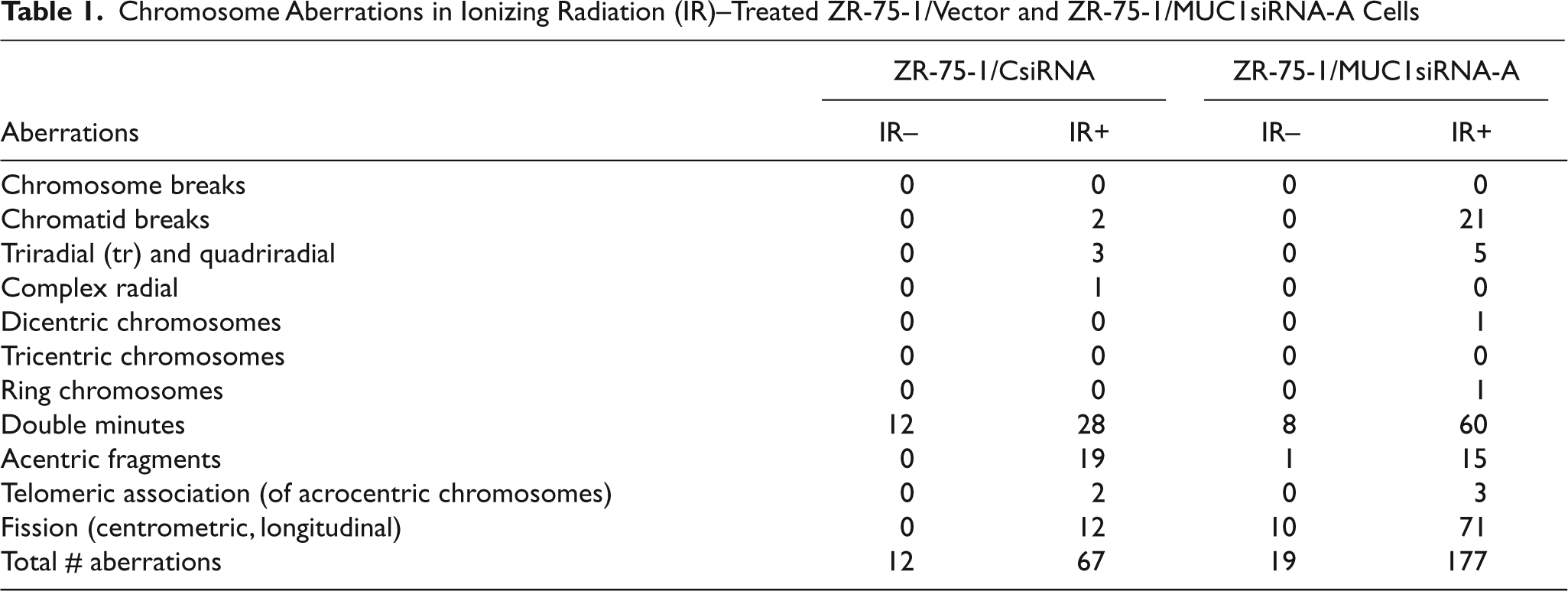
Phosphorylated H2AX accumulates at sites of DSBs. 40 γH2AX foci were detectable in the nuclei of irradiated ZR-75-1/vector cells (Fig. 5A). However, this response was decreased compared to that in irradiated ZR-75-1/MUC1siRNA cells (Fig. 5A). ATM-mediated phosphorylation of H2AX is also associated with activation of DNA repair. 2,40,41 In this regard, IR-induced γH2AX readily disappeared in ZR-75-1/vector cells as compared to the prolonged detection of γH2AX in ZR-75-1/MUC1siRNA cells (Fig. 5B). In concert with these results, analysis of γH2AX foci/cell over time demonstrated that MUC1 is associated with increased removal of γH2AX foci (Fig. 5C). To assess the effects of MUC1 on survival following IR treatment, we irradiated ZR-75-1 cells with 2 to 8 Gy. After 24 h, the cells were replated and assessed for colony formation at 3 weeks. IR treatment of ZR-75-1/vector cells had a partial effect on survival (Fig. 5D). By contrast, irradiation of the 2 ZR-75-1/MUC1siRNA clones was associated with substantial decreases in survival at doses of 2 and 4 Gy and the absence of colonies with treatment at 6 and 8 Gy IR (Fig. 5D). Analysis of chromosomes from the irradiated ZR-75-1 cells further demonstrated that silencing MUC1 is associated with an increase in aberrations, specifically chromatid breaks, double minutes, and fissions (Table 1). These findings collectively indicated that MUC1 promotes the rate of DNA DSB repair.

MUC1 promotes removal of ionizing radiation (IR)–induced γH2AX foci arrest and attenuates loss of survival. (
Chromosome Aberrations in Ionizing Radiation (IR)–Treated ZR-75-1/Vector and ZR-75-1/MUC1siRNA-A Cells
MUC1-CD is sufficient for promoting removal of IR-induced γH2AX foci
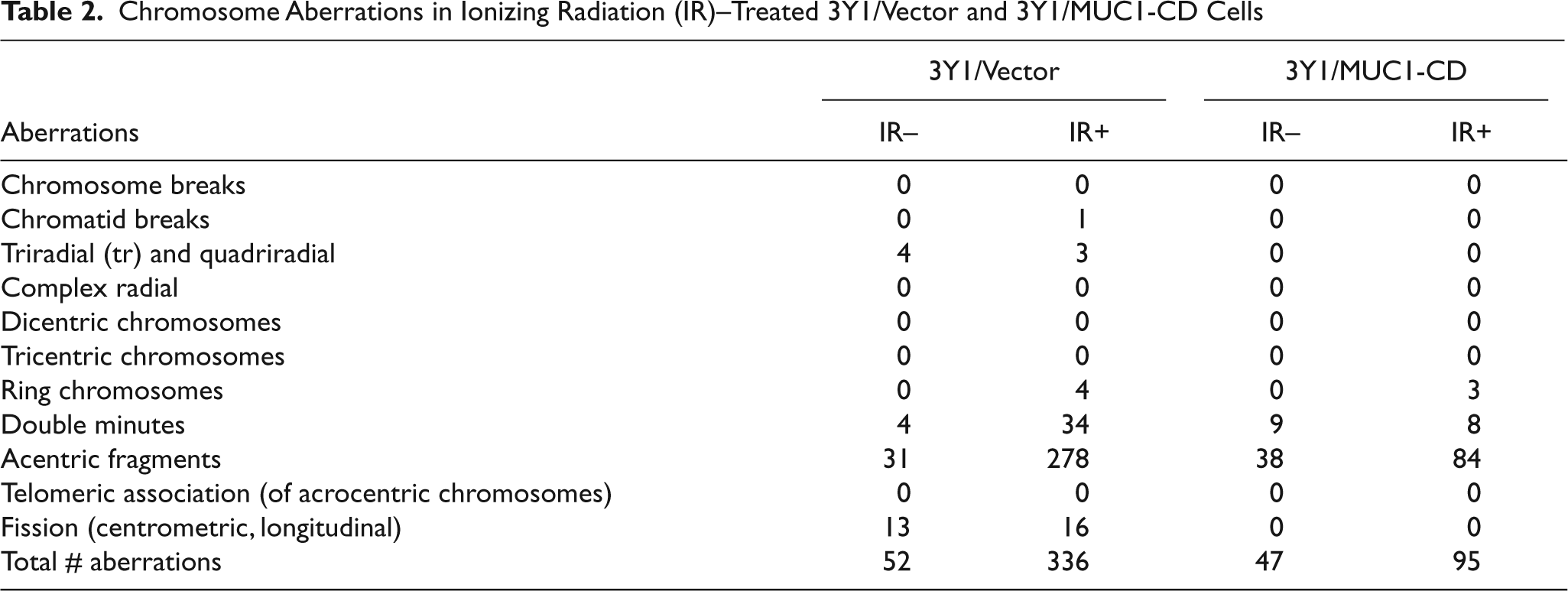
To determine whether MUC1-CD is sufficient for regulating H2AX phosphorylation, we studied rat 3Y1 fibroblasts that stably express an empty vector or MUC1-CD. 24 The results show that MUC1-CD attenuates H2AX phosphorylation in the response of 3Y1 cells to 2.5 and 10 Gy IR (Fig. 6A). Analysis of γH2AX foci/cell over time further demonstrated that MUC1-CD promotes removal of γH2AX foci (Fig. 6B). Moreover, expression of MUC1-CD in 3Y1 cells decreased the IR-induced arrest in G2-M phase as determined by phospho-histone H3 staining (Fig. 6C). To extend these results, the 3Y1 cells were treated with 7 Gy IR and then replated at different intervals after irradiation. The highest surviving fraction for the 3Y1/vector cells was at 4 h (Fig. 6D). Moreover, the highest surviving fraction for the two 3Y1/MUC1-CD clones was 24 h (Fig. 6D). In concert with the results obtained in ZR-75-1 cells, expression of MUC1-CD in 3Y1 cells decreased IR-induced chromosomal aberrations (Table 2). These findings indicate that MUC1-CD is sufficient to promote removal of γH2AX foci and attenuate chromosomal aberrations associated with DNA DSBs.
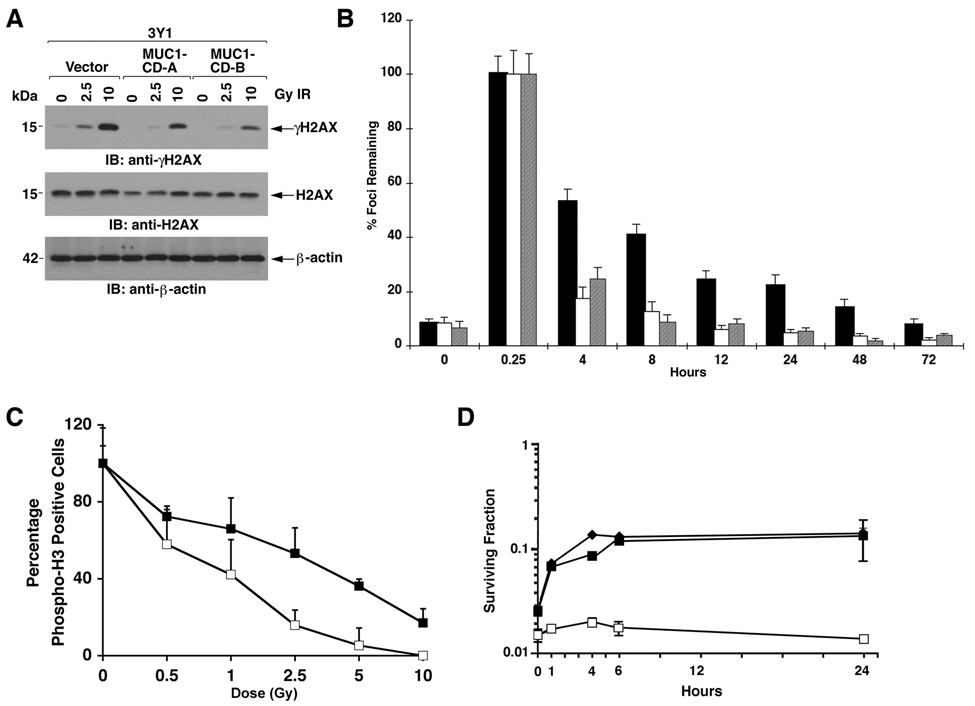
MUC1-CD is sufficient for promoting γH2AX removal. (
Chromosome Aberrations in Ionizing Radiation (IR)–Treated 3Y1/Vector and 3Y1/MUC1-CD Cells
Discussion
MUC1-C interacts directly with ATM
The present work demonstrates that the MUC1-C subunit associates with ATM in cells. The MUC1-C cytoplasmic domain binds directly to ATM HEAT repeats that are downstream to those identified for binding of NBS1. MUC1-C localizes to the nucleus in diverse cell types, 17,29,42,43 associates with transcription factors such as p53, and contributes to the regulation of gene activation. 27,30,44 Other work has shown that MUC1-C blocks death in the response to genotoxic anticancer agents. 31,35 However, to our knowledge, there has been no evidence to support involvement of MUC1-C in responses attributable to ATM function, such as DNA repair, activation of cell cycle checkpoints, or radiosensitivity. Nonetheless, the present results obtained from cells with stable silencing of endogenous MUC1 indicate that the MUC1-C oncoprotein affects levels of H2AX phosphorylation and survival in the response to IR. Similar results were obtained with cells that stably express MUC1-CD, indicating that the MUC1-C cytoplasmic domain is sufficient to mediate these effects. Notably, MUC1 had no apparent effect on IR-induced ATM activation (data not shown), indicating that MUC1-C affects downstream events that are mediated by the ATM-H2AX complex.
MUC1 promotes removal of IR-induced γH2AX foci
DNA DSBs can result in genomic instability if not subjected to adequate detection and repair. H2AX, an important component for optimal repair of these lesions, is phosphorylated on serines to form γH2AX foci at DNA DSB sites. 3 In this context, detection of γH2AX is used to assess the extent of initial DNA DSBs, and the rate of removal is employed as an indicator of DNA DSB repair. 3,15 The present studies demonstrate that silencing MUC1 in ZR-75-1 cells is associated with an increased intensity of γH2AX detection after IR treatment. In addition, in cells expressing endogenous MUC1, IR-induced γH2AX levels were substantially decreased by 4 h as compared to persistence of γH2AX at 24 h in the MUC1-silenced cells. The role of MUC1-CD in regulating γH2AX levels was confirmed in the IR response of 3Y1 cells. These results could have been attributable to a role for MUC1 in blocking ATM phosphorylation of H2AX in the response to IR. However, MUC1 had little if any effect on IR-induced activation of ATM and phosphorylation of other substrates, such as Chk2 (data not shown). The findings thus indicate that MUC1 promotes removal of IR-induced γH2AX, perhaps by promoting DNA DSB rejoining and thereby more rapid disappearance of γH2AX. Consistent with this model, MUC1 expression was clearly associated with more rapid kinetics of γH2AX removal following IR exposure. Moreover, survival in response to IR further indicated that MUC1 promotes the kinetics of γH2AX removal through DNA repair.
MUC1 protects against IR-induced cell death
Analysis of chromosomes from irradiated ZR-75-1 cells demonstrated that, in the absence of MUC1, there was an increase in chromatid breaks, double minutes, and fissions. Moreover, there was an increase in chromosome aberrations in irradiated 3Y1/vector cells, as compared to 3Y1/MUC1-CD cells, which were predominantly in the form of acentric fragments. In further support of a MUC1-mediated effect, we irradiated cells, and after 24 h, the cells were seeded and analyzed for clonogenic survival. In this assay, survival rates are greater for cells that are stimulated to grow immediately after irradiation, and the increase in survival corresponds to potentially lethal damage repair (PLDR). 45,46 The results obtained from both ZR-75-1 and 3Y1 cells demonstrate that MUC1 protects against IR-induced death. These findings obtained for cell cycle progression, chromosome aberrations, and survival collectively indicate that MUC1 expression is associated with increased repair of IR-induced DNA lesions. A striking aspect of these findings is that MUC1-C, a cell membrane-associated protein, interacts with the DNA repair machinery. In this regard, NEMO, an activator of the canonical NF-κB pathway and a MUC1-C binding partner, 26 associates with death receptors at the cell membrane and interacts with ATM. 47,48 However, the effects of the NEMO-ATM interaction on radiosensitivity are not known.
Why would MUC1 contribute to DNA repair?
MUC1 is a major component of a physical barrier that protects the epithelial cell layer from damage induced by diverse forms of stress, including ROS, which are encountered at the interface with the external environment. In the epithelial stress response, damage induces the disruption of tight junctions, loss of polarity, and activation of a repair and survival program. 49 The available evidence indicates that MUC1 contributes to this repair and survival program through nuclear targeting of the MUC1-C subunit. 27-29,35 Thus, in the stress response of normal epithelial cells, nuclear MUC1-C could transiently protect against damage-induced DNA lesions. By contrast, with transformation, MUC1-C is constitutively expressed in the nucleus. 27-29,35 Irreversible nuclear targeting of MUC1-C in carcinoma cells could therefore confer a phenotype, as found in ZR-75-1 cells, that constitutively promotes repair of DNA lesions. In this regard, other studies have shown that MUC1 protects against death induced by ROS that, like IR, also induce DNA DSBs. 36-38 Radiotherapy is widely used as a treatment modality for breast cancer; however, for patients who develop recurrence of their disease, the mechanisms responsible for radioresistance are not well understood. MUC1-induced repair of DNA lesions could protect against genomic instability but also contribute to protection of tumors against treatment with IR and other genotoxic anticancer agents. Therefore, human breast tumors that overexpress MUC1 may have a survival advantage to genotoxic stress by exploiting mechanisms that evolved for the repair of damaged epithelia. In that context, recent studies have demonstrated that direct targeting of MUC1-C oligomerization in breast cancer cells blocks nuclear localization of MUC1-C and activates the DNA damage response. 50 The MUC1-C inhibitor could thus be effective in potentiating the therapeutic response of radiation in the treatment of breast cancers.
Materials and Methods
Cell culture
ZR-75-1/vector and ZR-75-1/MUC1siRNA cells 31 were grown in RPMI1640 medium containing 10% heat-inactivated fetal bovine serum (HI-FBS), 100 units/mL penicillin, 100 µg/mL streptomycin, and 2 mM L-glutamine. 293 embryonal kidney cells, rat 3Y1/vector, and 3Y1/MUC1-CD fibroblasts 24 were cultured in Dulbecco’s modified Eagle’s medium with 10% HI-FBS, antibiotics, and L-glutamine. Cells were irradiated at room temperature with a γ-ray source (Cs173; Gamma Cell 1000; Atomic Energy of Canada, Ottawa, Canada) at a fixed dose of 0.76 Gy/min.
Immunoprecipitation and immunoblotting
Lysates were immunoprecipitated with anti-MUC1-C (Ab-5; Neomarkers, Fremont, CA), anti-ATM, anti-Myc (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-Flag (M2; Sigma, St. Louis, MO). The precipitates and lysates not subjected to immunoprecipitation were immunoblotted with anti-MUC1-C, anti-ATM, anti-GFP (BD Biosciences Clontech, Palo Alto, CA), anti-ATM-phospho-Ser-1981 (Rockland, Gilbertsville, PA), anti-β-actin (Sigma), and anti-γH2AX and anti-H2AX (Cell Signaling Technology, Beverly, MA). Immunocomplexes were detected with horseradish peroxidase–conjugated secondary antibodies and enhanced chemiluminescence (ECL; Amersham Biosciences, Piscataway, NJ).
In vitro binding assays
GST and GST-ATM fusion proteins bound to glutathione beads were incubated with cell lysates or purified His-MUC1-CD in binding buffer 24 for 4 h at 4°C. Adsorbates to the beads were washed 6 times and subjected to immunoblot analysis.
Immunofluorescence
Cells in culture were fixed in 3% paraformaldehyde/phosphate-buffered saline (PBS) for 10 min and permeabilized in 0.5% Triton X-100 for 5 min. Incubations with primary antibody and with Rhodamine Red-X secondary antibody were each performed for 30 min at 37°C. Nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI). The stained cells were analyzed under a fluorescence microscope.
Flow cytometry
Cells were harvested, washed with PBS, fixed with 80% alcohol, and stained with PI/RNase staining buffer (BD Biosciences PharMingen, San Diego, CA) for 60 min at 4°C in the dark. DNA content was analyzed by flow cytometry.
Cell cycle G2/M arrest assay
Cells seeded in 6-well plates with coverslips were irradiated, incubated for 1 h, fixed, and stained with anti-phospho-histone H3 antibody (Cell Signaling Technology) and DAPI. Cells in mitosis were counted and expressed as a percentage of the control untreated mitotic cell number.
Clonogenic survival assays
Cells were treated with IR, incubated for 24 h, and then plated at 500 to 5000 cells/well in 6-well plates. After 1 to 3 weeks, colonies were stained with crystal violet and counted manually. The surviving fraction was calculated as (number of colonies formed)/(number of cells plated × plating efficiency).
Footnotes
Acknowledgements
The authors thank Dr. M. Kastan for providing the pcDNA3-Flag-ATM plasmid.
Dr. Kharbanda is an employee of Genus Oncology. Dr. Kufe is a founder of Genus Oncology and a consultant to the company. The other authors declare no potential conflicts of interest.
This work was supported by the National Cancer Institute (grant numbers CA97098, CA42802), the National Natural Science Foundation of China (grant numbers 30671169, 30871363), and the Shanghai Pujiang Program (grant number 07pj14065).