
Abstract
Wilson’s Disease (WD) manifests with systemic and neuropsychiatric symptoms, caused by an ATP7B genetic mutation, leading to an accumulation of copper. Presentations are diverse and the diagnosis should be considered in anyone under 50 with a new onset movement disorder. Early recognition and treatment can limit morbidity. While liver transplantation (LT) is recommended in WD patients with hepatic failure, its use for pure neurologic indication remains controversial. We present a patient who failed medical management and underwent LT for pure neurologic indications. Subsequent neurologic symptom improvement supports the use of LT for patients with pure neurologic manifestations of WD.
Keywords
Case Report
A 22-year-old woman developed bilateral upper extremity action tremor and presented to an outside hospital where she was diagnosed with essential tremor and started on propranolol. She had no family history of tremor or other movement disorders. As hyperkinetic movements developed she underwent further workup. Laboratory evaluations detected low serum ceruloplasmin (7.5, normal range 19.0-39.0 mg/dL), elevated urine copper (79 μg/24 hr, normal range 3-35 μg/24 hr), and elevated copper/creatinine ratio (136, normal range 0-49). MRI findings were consistent with Wilson’s Disease (WD) (Figure 1). She was found to have Kayser Fleischer rings in both eyes; no sunflower cataracts were noted. Trientine and zinc were initiated, but she suffered neurologic deterioration despite (or due to) this treatment.
1
Pre-transplant MRI findings. Axial T1 (1A-B) and T2 FLAIR (1C-D) showed hyperintensity of basal ganglia and thalamus in addition to the characteristic “face of the giant panda” sign in the midbrain.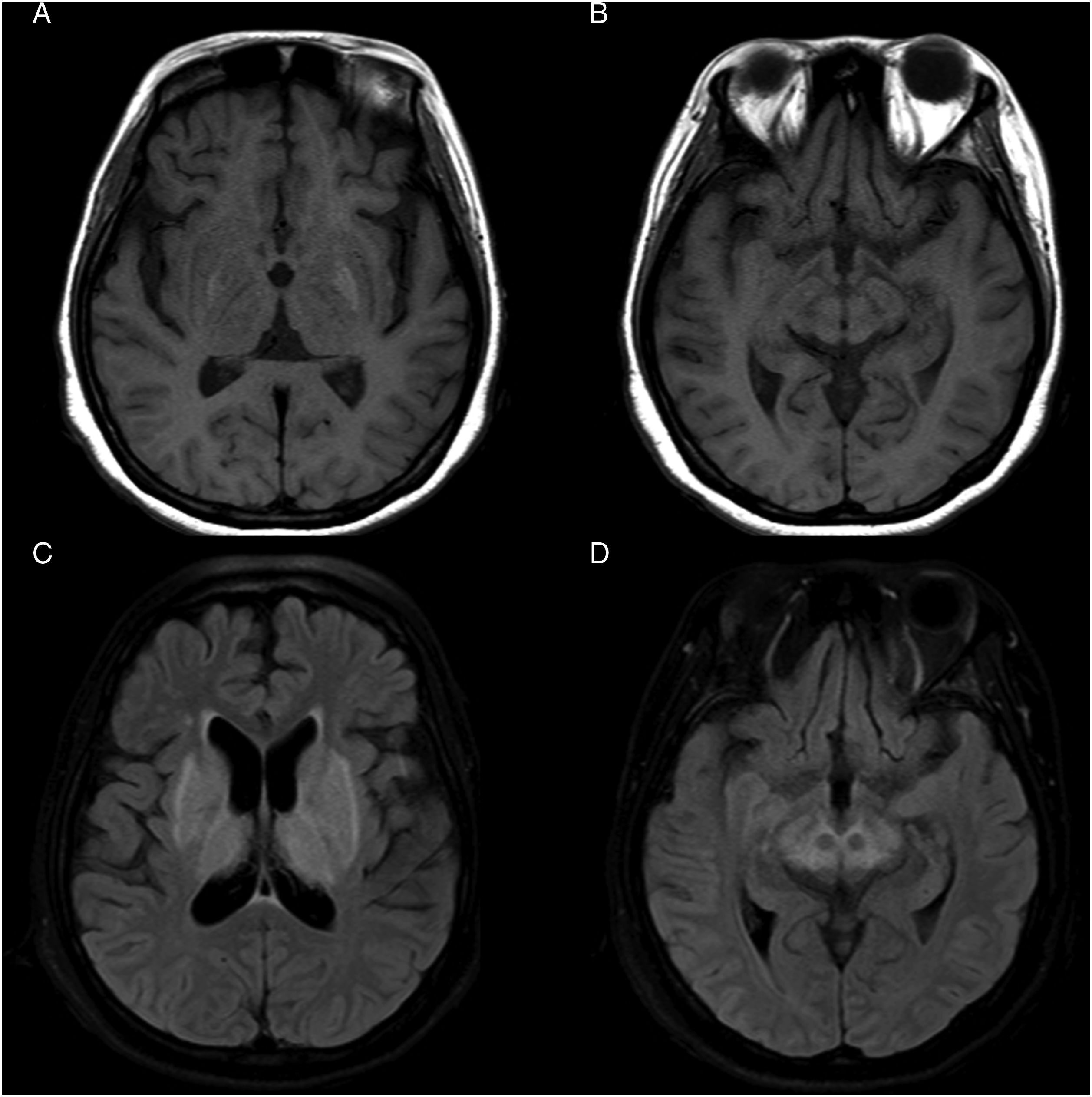
Over the subsequent 7 months, her gait and balance worsened and she developed encephalopathy and more severe bilateral appendicular hyperkinetic movements (Video 1). The patient’s decline culminated in her presentation to our hospital in status dystonicus, which required intravenous sedation. Labs on initial presentation to our hospital were as follows: serum copper 25 (normal range 80 - 158 μg/dL) and 24-hour urine copper 1211 μg/24 hr (normal range 3-35), suggesting appropriate chelation.
Although liver dysfunction was minimal on presentation (AST 122, ALT 366, ALP 185, total bilirubin .3, Albumin 2.4, INR 1.1), she underwent orthotopic liver transplantation to maintain copper balance and to avoid the need for oral chelation. In the postoperative period, symptomatic treatment for dystonia and spasticity included clonazepam, baclofen, trihexyphenidyl, carbidopa-levodopa, tetrabenazine, and botulinum toxin injections. The patient’s pre-transplantation Unified Wilson Disease Rating Scale (UWDRS) score was 157. Six months post-transplantation, her UWDRS score had improved to 74 (Video 2). Her Modified Rankin Scale (MRS) improved from 5 to 4. At last assessment, she was able to self-propel in a wheelchair, speak intelligibly, and use her arms for activities of daily living. Her independent gait was limited by persistent ataxia. Mild improvements in T2 hyperintensity were noted on repeat MRI (Figure 2C, D). Eight months after transplant, the patient had weaned down her symptomatic treatments, and she remained off zinc and trientine. Post-transplant MRI findings. Axial T1 (1A-B) and T2 FLAIR (1C-D) showed improved appearance of the hyperintensities of basal ganglia and thalamus.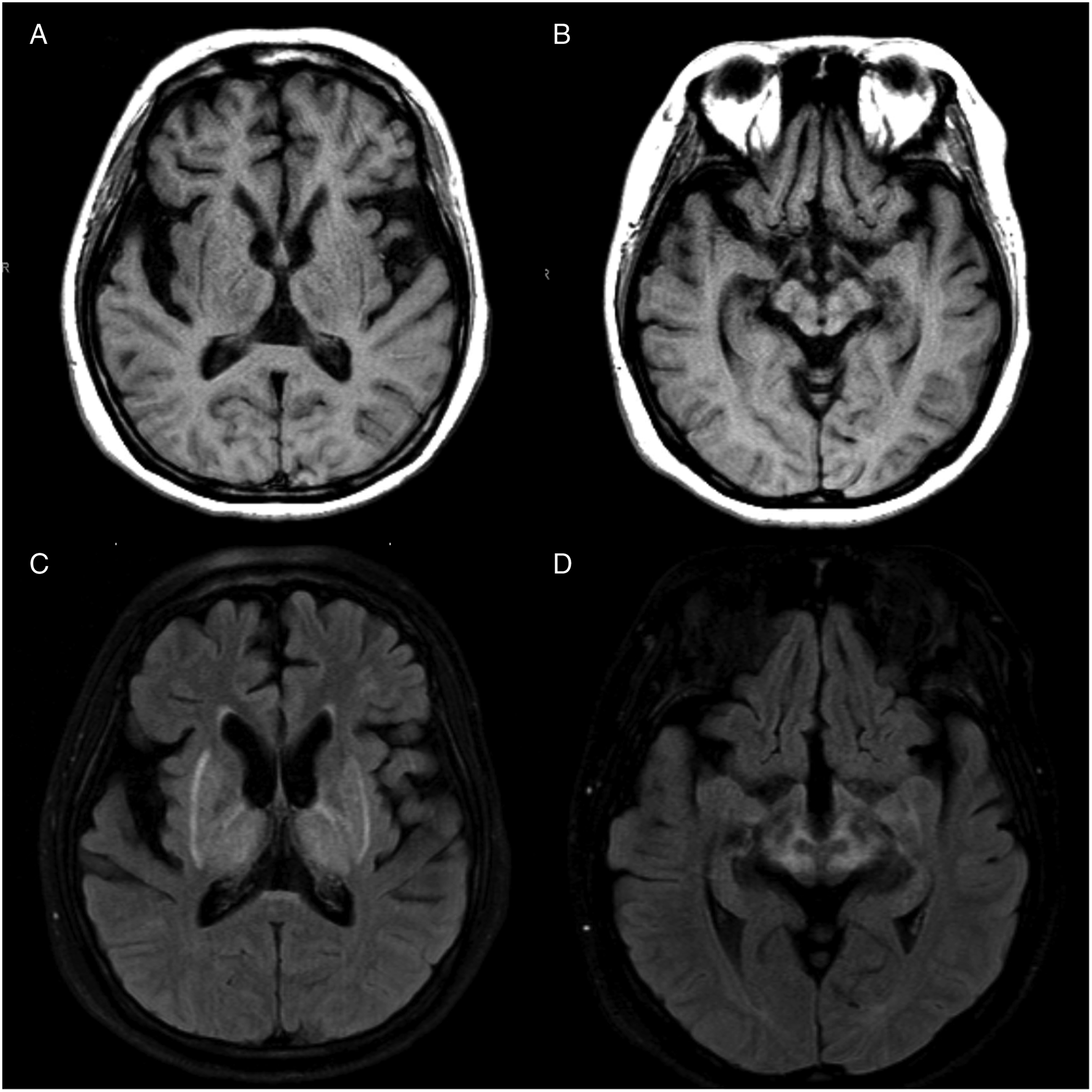
Discussion
Wilson’s Disease (WD) is a disorder of copper metabolism with broad systemic and neuropsychiatric manifestations. WD is an autosomal recessive condition that affects 1 in every 30,000 individuals. Most commonly, patients have two abnormal copies of the ATP7B gene, which encodes for a cation transport enzyme that serves two functions. First, ATP7B enables hepatocytes to link copper with ceruloplasmin so that the metal can safely travel in systemic circulation. Second, ATP7B allows for the excretion of excess copper via bile. Urinary excretion of copper is negligible, and copper excretion through bile enables clearance of the 1-2 mg of daily dietary copper intake. Without a properly restrictive diet and appropriate treatment, patients accumulate copper in their liver. This leads to oxidative stress and liver dysfunction, explaining early presentation in the first decades of life and histological findings that resemble chronic hepatitis. As disease progresses in the second and third decade, excess copper spills out of the liver and into the bloodstream where, unbound, it precipitates. This copper deposits in the kidneys and the brain, where it has a predilection for the basal ganglia. 2
Wilson’s disease usually presents first with hepatic symptoms (fatigue, nausea, jaundice, ascites, and hepatosplenomegaly) followed by neuropsychiatric involvement, unless the patient has a cryptogenic course without any hepatic symptoms, and therefore presents with neurologic or psychiatric conditions. Neuropsychiatric symptoms have myriad presentations which can occur in isolation or in combination, including dysarthria, tremor, sialorrhea, dystonia, ataxia, parkinsonism, mood disorders, and psychosis. 3 Myoclonus, chorea, and seizures are less common potential manifestations. There are two ophthalmic signs also common in WD. The deposition of copper in the eye can lead to a golden-green discoloration of the periphery of the cornea, within Descemet’s membrane, called Kayser-Fleischer rings. The second ophthalmic sign of WD is sunflower cataracts, which is the cuprous opacification of the center of the lens. WD can also cause hemolytic anemia and arthritis.
Neuroimaging in WD patients with neurological dysfunction is often abnormal, and MRI can show bilateral midbrain and pontine T2 signal changes which create the “face of the giant panda” and the “miniature panda” signs, respectively. When seen together, the pareidolia has been called the double panda sign. 4 T2 hyperintensity can involve the thalamus and basal ganglia as well. Copper deposits are visualized as T1 hyperintensities. Global atrophy can be seen. 2 Morphological abnormalities have been detected in deep structures, particularly the caudate, putamen, and thalamus, but also the globus pallidus, amygdala, red nucleus, and substantia nigra. 2 In addition to imaging, a Leipzig score can help diagnose WD, with or without genetic testing. This scoring system incorporates presence of KF rings, severity of neurologic symptoms, serum ceruloplasmin, liver and urinary copper, presence of Coombs-negative hemolytic anemia, and chromosomal mutation analysis; a score greater than 4 establishes a diagnosis of WD. Patients can be screened by collecting a 24-hour urine copper. A ceruloplasmin level blood test is a supportive testing approach but offers low diagnostic value in isolation given its relatively low sensitivity, 84%. 5
It is important to recognize the myriad initial presentations of Wilson’s Disease and that we maintain a low threshold to evaluate for WD in younger patients with movement disorders. Atypical presentations of WD are uncommon but are relevant and should be considered to make the diagnosis. The French Wilson’s Disease Registry reported 8% of their cohort (45) patient were diagnosed after age 40 years (median age 49). 6 Many patients with these later diagnoses had predominantly neurologic presentations with potentially isolated long-lasting symptoms (including writer’s cramp, cervical dystonia, and functional neurologic disorders—tremor and myoclonus). There was a notable absence of KF rings and MRIs were unremarkable. Screening at-risk family members, particularly pre-manifest siblings of a proband, offers the potential for pre-symptomatic diagnosis and prompt treatment, hopefully minimizing morbidity.
Treatment of WD focuses on inhibiting copper absorption with zinc salts, limiting dietary copper, and increasing copper excretion with chelating agents (penicillamine, trientine, and tetrathiomolybdate). 7 Interval monitoring of serum and urine copper levels (and/or zinc levels) ensures therapeutic efficacy, treatment compliance, and avoids over-chelation and the complications of copper deficiency. However, chelation itself can present significant risks to patients, most notably bulbar symptoms and exacerbation of neurodegenerative decline. 1 Penicillamine and trientine have been reported to cause neurologic worsening in up to 50% of WD patients, potentially by mobilizing stored copper, thus further elevating brain copper levels. 1 Clinical markers that have been associated with post treatment decline include: initial neurologic presentation (as opposed to hepatic presentation), higher UWDRS parts II and III scores, thalamic and brainstem pathology on MRI, and treatment with dopamine antagonists. 8 Additionally, elevated serum neurofilament light chain levels have been reported as possibly predict this early neurologic deterioration. 9 In a single center retrospective audit, penicillamine also caused significant hematologic and renal adverse events, with significant intolerance noted in 28 of the 112 patients studied. 10 There is an ongoing phase 3 study for an adeno-associated virus-mediated gene transfer therapy for WD, which depending on the outcome could change this therapeutic conversation drastically. 11 This study is planned to complete in 2031.
Currently, liver transplantation (LT) is recommended for WD patients with hepatic failure.12,13 However, the efficacy of LT to treat patients with pure neurologic indication is unresolved. A systematic review of LT for neurologic WD showed a discrepancy between older and newer studies, with newer studies reporting better results of neurologic improvement and better survival rates. 8 A retrospective cohort study in 2020 looked at eighteen French patients ranging between 16-20.8 years of age who were resistant to decoppering agents and who underwent LT as rescue therapy for pure neurologic indications. Eight of these patients experienced a statistically significant improvement in their Unified Wilson’s Disease Rating Scale (UWDRS) score, decreasing on average by 78%. 14 Brain MRI scores and Kayser-Fleischer rings also improved. Whether due to the intolerance or ineffectiveness of such chelating agents, neuropsychiatric WD patients should be considered for LT, traditionally reserved for patients with hepatic failure.
We present the case of a patient who underwent liver transplant for pure neurologic indications with subsequent neurologic symptom improvement after symptomatic worsening with chelation therapy. Essential tremor was a “diagnostic placeholder” in our patient until other neurologic symptoms developed. 15 Delay in diagnosis may have negatively impacted this patient’s outcome, including increasing the risk for chelation-related neurologic deterioration. Our case adds to the growing body of evidence that supports the beneficial role of liver transplant in the treatment algorithm for neurologic WD, particularly for patients with pure neurologic manifestations of WD who fail medical management. LT may be a reasonable therapeutic option when chelation fails to show benefit or is not well tolerated. Further studies are needed to determine demographic predictors of post-LT improvement in neurologic WD.
Supplemental Material
Supplemental Material
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.