
Abstract
Background
Infantile myofibromatosis (IM) is a disorder characterized by proliferation of benign myofibroblastic tumors that typically manifest as solitary or multiple nodules in the skin, muscle, bone, subcutaneous tissues, and visceral organs and can pose significant morbidity and mortality risks, particularly in cases involving visceral organs or causing functional impairment. These soft tissue lesions are the most prevalent benign fibrous tumors that present before age two and can undergo spontaneous regression or are amenable to surgical resection.
Case
A preterm, male infant was born via Caesarean section to a mother with a trichorionic, triamniotic pregnancy following preterm labor. Within the first week of life, several well-circumscribed, smooth, non-tender, and soft nodules with some mobility were noticed along the border of the ribs, across the trunk, back, and lower extremities. Ultrasound imaging confirmed well-circumscribed hypoechoic, intramuscular nodules, and biopsy evaluation showed atypical spindle cell proliferation. The biopsied lesion was PDGFRB-mutated on molecular genetic studies, confirming a diagnosis of myofibromatosis. The infant developed mixed lytic and sclerotic deformities of a variety of bones, necessitating treatment given disease progression.
Conclusion
Successful clinical management with low-dose metronomic chemotherapy (methotrexate and vinblastine) is possible and can treat extensive disease, as seen in our patient.
Background
Infantile myofibromatosis (IM) is a disorder characterized by the proliferation of benign myofibroblastic tumors that typically manifest as solitary or multiple nodules and can involve the skin, muscle, bone, subcutaneous tissues, and visceral organs. 1 Primarily affecting infants and young children, the condition was first described by Stout in 1954 as “congenital generalized fibromatosis.” 2 Although it is rare with a documented incidence of one in 150,000 live births, IM is the most prevalent benign fibrous tumors during the initial year of life and generally presents before age two. 3 IM has been classified into three main types: (i) solitary IM, (ii) multicentric IM with visceral involvement, and (iii) multicentric IM without visceral involvement. 3 Diagnosis is primarily achieved through clinical examination, imaging studies, and histopathological analysis, although challenges may arise due to its heterogeneous presentation and rarity. The etiology of IM remains unclear but is believed to have a genetic component; both autosomal dominant and recessive inheritance patterns have been described in the literature. 4 In particular, PDGFRB gene mutations have been identified as a common association with cases of IM.3,4 Herein, we present a preterm male infant diagnosed with multicentric IM without visceral involvement and PDGFRB mutation positive requiring prolonged chemotherapy.
Case presentation
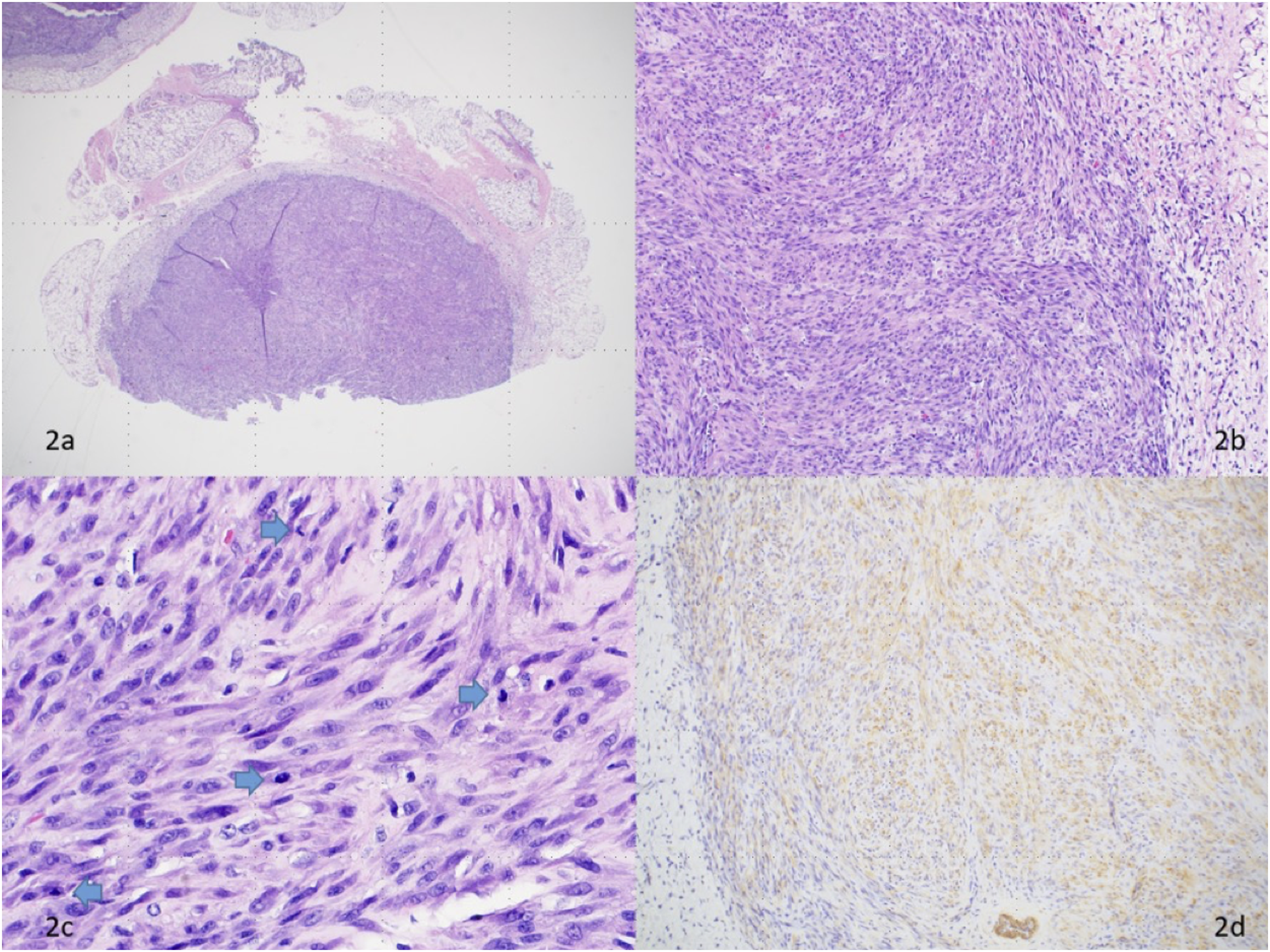
A preterm 28 3/7 weeks, Caucasian, male infant was born via Caesarean section to a 27-year-old G1P3 mother with a trichorionic, triamniotic pregnancy following preterm labor. Within the first week of life, several well-circumscribed, smooth, non-tender, soft nodules with some mobility were noticed along the border of the ribs, across the trunk, back, and lower extremities [Figure 1]. Ultrasound imaging at 2 weeks of life confirmed well-circumscribed hypoechoic, intramuscular nodules with internal echogenic foci and internal vascularity on Doppler. Biopsy evaluation at 2 and 7 weeks of life showed an atypical spindle cell proliferation [Figure 2]. The biopsied lesion was PDGFRB-mutated on molecular genetic studies, raising considerations for myopericytoma, myofibroma, and fibrosarcoma as potential differential diagnoses. There were two mutation variants of clinical significance detected, PDGFRB p.N666 K missense and PDGFRB p.V568_I569del novel in-frame deletion. The p.N666 K variant alters the kinase domain of PDGFRB and has been previously observed in cases of both myopericytoma and myofibroma.5,6 Gain-of-function germline and somatic mutations in the kinase and juxtamembrane domains of PDGFRB have been commonly found in both familial and sporadic infantile myofibromatosis.5,7 The assay was unable to distinguish the PDGFRB mutations between germline or somatic variants. The novel p.V568_I569del variant in the juxtamembrane domain appears to be previously unreported, but this domain is known to be a mutation hotspot for PDGFRB in myofibroma.4,6–8 Several mutations nearby have been documented in infantile myofibromatosis, often co-occurring with PDGFRB kinase domain mutations in myofibroma lesions.
9
A separate test for ETV6/NTRK3 associated with chromosome 12 and 15 translocation was negative for the fusion that is characteristic of infantile fibrosarcoma. Together with the histopathologic findings, molecular genetic studies, and clinical context, the results were supportive of a diagnosis of multifocal infantile myofibromatosis. Well-circumscribed, smooth, non-tender, soft nodules with some mobility along the border of the ribs, across the trunk, back, and extremities. Histopathologic images of one representative subcutaneous nodule: (a) shows a low-power image with a well-circumscribed proliferation of spindle and plump myoid cells [H&E, 20x]. (b) shows the spindle cells arranged in short interlacing fascicles [H&E, 100x]. (c) is a high-power view showing tumor cells with slightly pleomorphic cigar-shaped vesicular nuclei and frequent mitoses (blue arrows) [H&E, 400x]. (d) shows lesional cells staining with smooth muscle actin (SMA) immunohistochemical stain [SMA, 100x].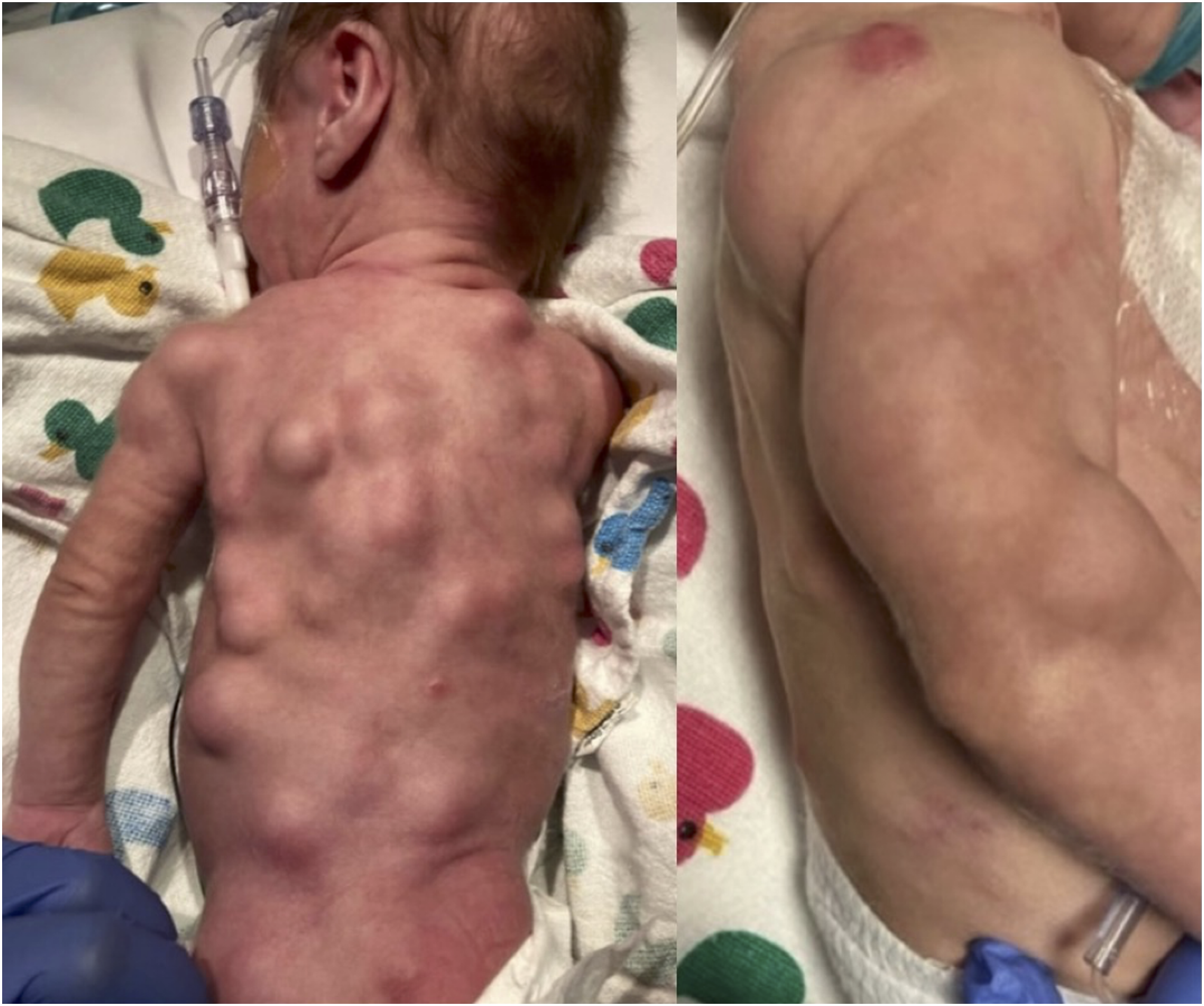
Imaging evaluation included a full body MRI and brain MRI depicting metastatic sarcomatous disease without intraosseous, visceral, or intracranial involvement. Innumerable circumscribed, ovoid heterogeneous lesions isolated to musculature and superficial subcutaneous tissues were found throughout the body. Brain MRI was significant for numerous nodular lesions present in the masticator space, mandible, and neck with evidence of a prominent mass in the left temporal fossa [Figure 3]. Brain MRI depicting a prominent hyperintense nodule in the left temporal fossa. No intracranial lesions were seen on the MRI.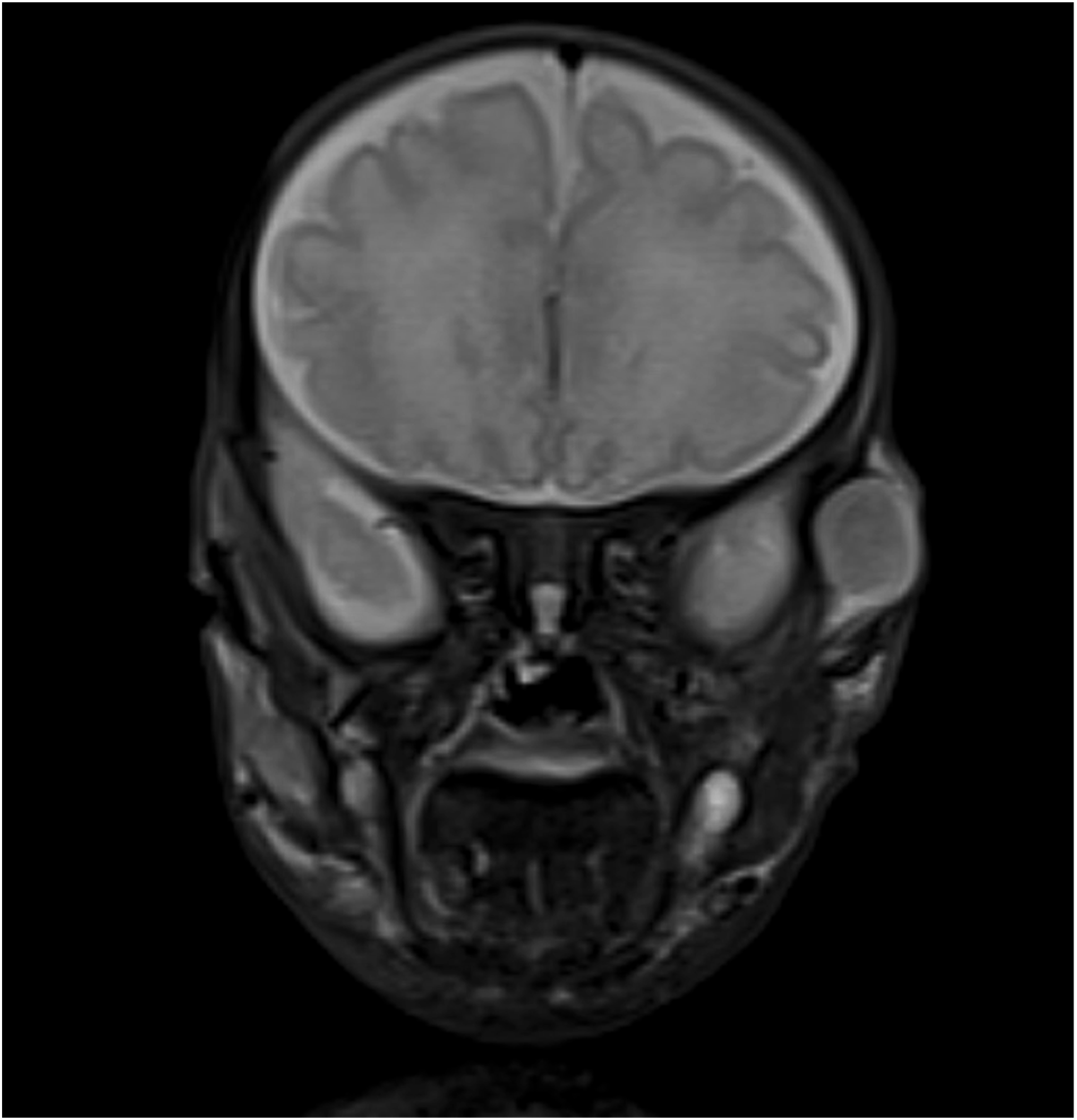
Given the natural history of spontaneous regression and the difficulties of chemotherapy administration in the preterm infant, we monitored progression of the disease with clinical exam. Unfortunately, the disease progressed, and the infant developed mixed lytic and sclerotic deformities of a variety of bones, necessitating treatment. Given the number of lesions, surgical resection was not a possibility, and the infant was treated with low-dose metronomic weekly methotrexate (1 mg/kg) and vinblastine (0.2 mg/kg) with careful monitoring for tumor lysis syndrome and adverse medication effects. The patient was discharged from the neonatal intensive care unit (NICU) at 3 months of life with interval diminishment in the size of nodules. Weekly chemotherapy is ongoing at an outpatient infusion center for a tentative plan of 12 months of treatment with routine monitoring by Pediatric Hematology-Oncology.
Discussion
Initial differential diagnosis of these lesions included soft tissue tumors, lipomas, and hemangiomas. The physical characteristics of the lesions being non-tender, soft, moveable, and generalized in distribution were consistent with lipomas. Some of the initial lesions had a violaceous hue thereby raising the possibility of them being hemangiomas. However, with the varied distribution and rapid multiplication of the lesions, a biopsy and histological evaluation were deemed necessary. Based on tissue involvement, IM is classified into three main types: (i) solitary IM, (ii) multicentric IM with visceral involvement, and (iii) multicentric IM without visceral involvement. Depending on the type of IM, management typically involves a multidisciplinary approach and can encompass a combination of surveillance, surgical resection, or medical therapy.
Although histologically benign, IM can pose significant morbidity and mortality risks, particularly in cases involving visceral organs or causing functional impairment. 10 Our patient had no visceral involvement which reduced his risk of morbidity and mortality. Long-term prognosis varies widely, with some lesions spontaneously regressing while others may persist or recur,1,10 necessitating vigilant monitoring and individualized management strategies tailored to each patient’s clinical presentation. Surgical excision is reserved for cases with compromised vital functions. However, when the lesions are as extensive as seen in our patient, they can be treated with chemotherapy (interferon alfa, vincristine-actinomycin D-cyclophosphamide, and vinblastine-methotrexate).
Currently, after 3 months of chemotherapy, our patient has had near complete regression of all the lesions. The underlying mechanism of tumor regression and growth remains unknown, but it has been suggested to be related to angiogenic stimulation and regression, both triggered by basic Fibroblast Growth Factor (bFGF).4,11
Despite its low incidence rate, IM poses significant diagnostic and therapeutic challenges, emphasizing the importance of continued research efforts to improve understanding, diagnosis, and management of this condition. The PDGFRB mutation is known to cause autosomal dominant IM but can easily be missed with its incomplete penetrance and variable expressivity.
Conclusion
Multifocal infantile myofibromatosis can pose significant morbidity and mortality. Successful clinical management with low-dose chemotherapy is possible and is tolerated in premature infants with diligent clinical management from specialized care teams.
Statements and declarations
Footnotes
Author contributions
Drs. Lee, Amin, Chikwava, Smith, and Fernandes have disclosed no financial relationships relevant to this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.