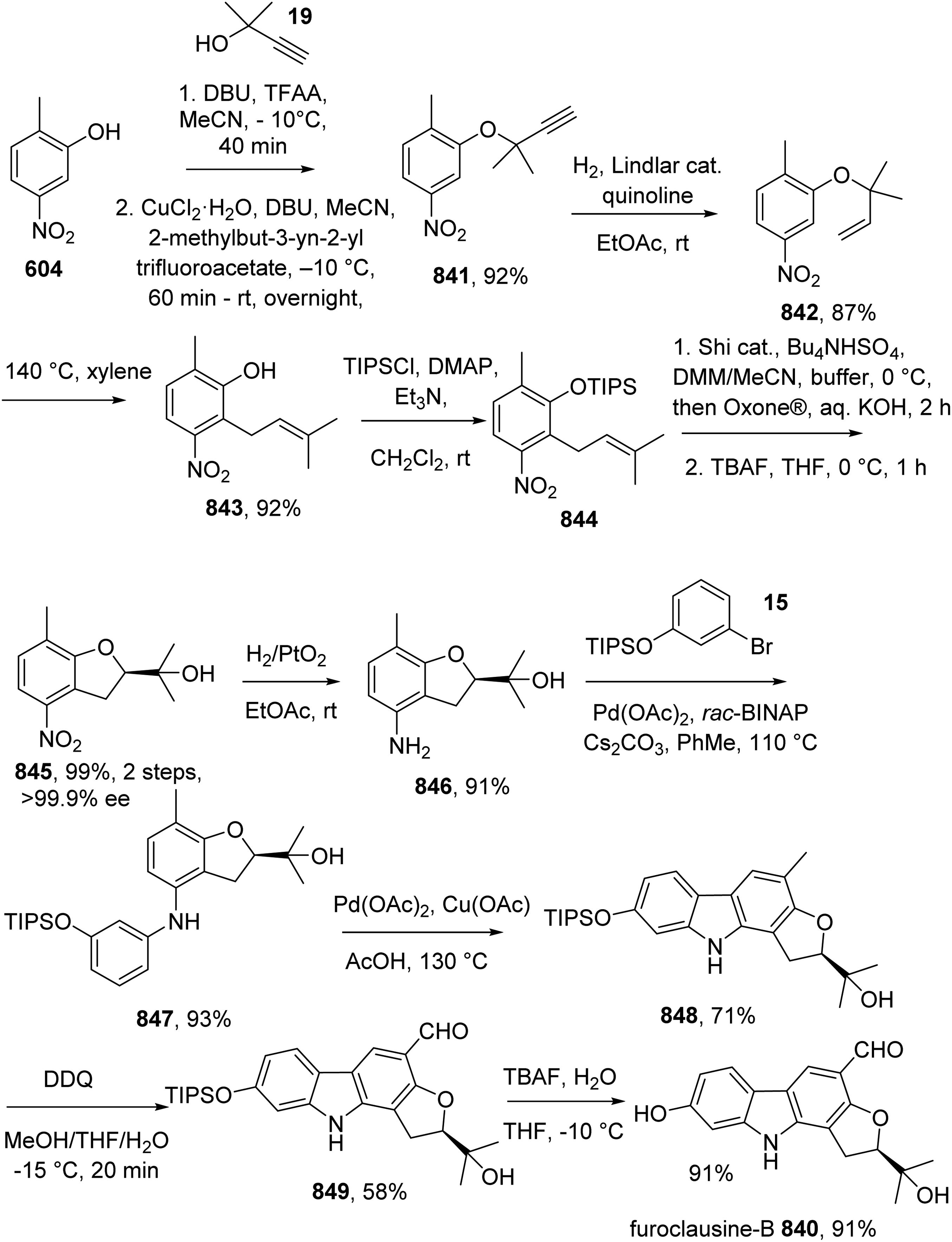
Carbazoles alkaloids are tricyclic aromatic heterocyclic organic compounds, consisting of a central pyrrole ring fused with two benzene rings. Carbazole derivatives are an important class of nitrogen-containing heterocyclic compounds that are widely found in nature and in synthetic source. These heterocyclic compounds have a broad range of bioactivities and thus, the synthesis of carbazole alakaloid has atracted intensive research interest of chemists. However, review articles on total synthesis of natural carbazole alkaloids in the literature are very limited and quite outdated. This review provides a comprehensive analysis of studies on the synthesis of natural carbazole alkaloids since 2014. More than 90 studies have been summarized. References have been collected from various resources such as Google Scholar, SciFinder, and PubMed. Especially, we tried to focus on total synthesis studies. The article might be useful for chemists who works in synthesis of heterocycles or drug chemistry.
Carbazoles alkaloids are tricyclic aromatic heterocyclic organic compounds, consisting of a central pyrrole ring fused with two benzene rings. Carbazole derivatives are an important class of nitrogen-containing heterocyclic compounds that are widely found in nature1 and in synthetic source.2 These heterocycles possess a wide range of bioactivities2,3 and some of them have been marketed as drugs for the treatment of various diseases (Figure 1). For example, ondansetron is a drug used to prevent nausea and vomiting, midostaurin is a multi-targeted protein kinase inhibitor, and cardevilol is a β-blocker. The diverse bioactivities of carbazoles such as antimicrobial, anti-inflammatory, anticancer, neuroprotective, and antidiabetic activities have been well documented in the literature.2,3 Review articles on synthesis of carbazole based compounds have also well described.4,5 However, review articles on total synthesis of natural carbazole alkaloids in the literature are very limited and quite outdated.6,7 In this report, we will demonstrate a comprehensive review of natural carbazole alkaloids syntheses dating back to 2014.
Some Drugs with Carbazole skeleton.
Synthesis of Simple Natural Carbazole Alkaloids
Synthesis of Simple Natural Carbazole Alkaloids by the Knölker Group
The Knölker group has made a great contribution to the total syntheses of carbazole alkaloids and a numerous number of studies on total syntheses of natural carbazole alkaloids have been published. The group also has been well-known for isolation, bioactivities of natural carbazole alkaloids as well as construction of carbazole skeleton.2,6
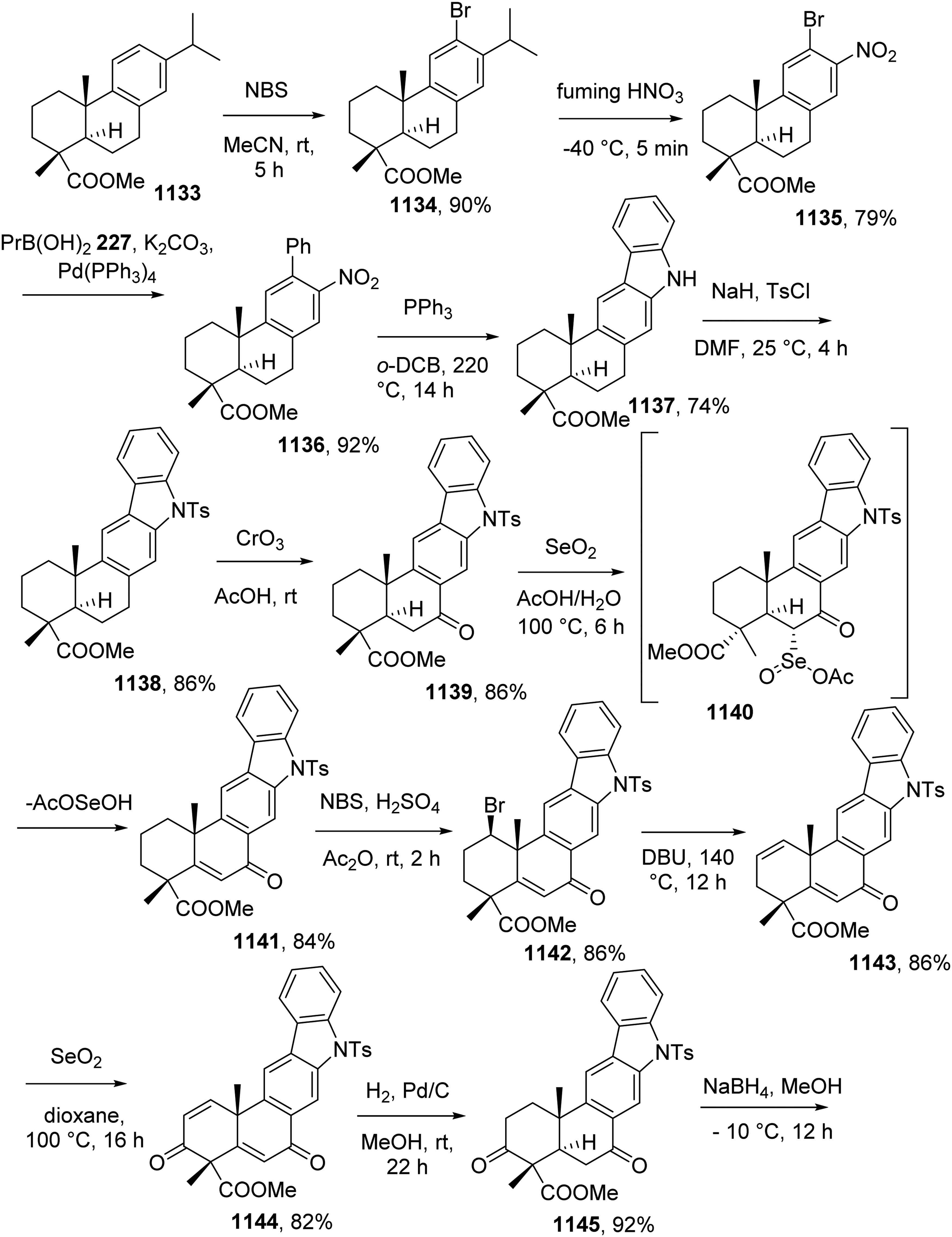
In 2014, the group accomplished the total syntheses of a series of natural carbazole alkaloids including heptaphylline 1, 7-hydroxyheptaphylline 2, 7-methoxyheptaphylline 3, mukoenine-B 4 (clausenatine-A), euchrestine-A 5, and isomurrayafoline-B 6 based on the DIBAL-H promoted reductive pyran ring opening of dialkylpyrano[3,2-a]carbazoles.8
The Buchwald-Hartwig coupling of the arylamine 7 and iodobenzene 8 gave diaryl amine 9, which was converted to carbazole 10 by palladium(II)-catalyzed oxidative cyclization. Acid-promoted cleavage of the methyl ether in 11 provided pyranocabazole 12, which was transformed into pyrano[3,2-a]carbazole 13 by a sequence of dimethylpropargyl ether formation and thermally induced rearrangement. This pyranocarbazol was reduced to heptaphylline 1 by DIBAL-H in 75% yield. For the preparation of 7-hydroxyheptaphylline 2, Buchwald-Hartwig amination reaction between arylamine 14 and bromide 15 led to diaryl amine 16, which underwent palladium(II)-catalyzed oxidative cyclization to generate carbazole 17. Benzyl deprotection of 17 resulted in 18, which was transformed to pyrano[3,2-a]carbazole 21 by annulation of the pyran ring via Godfrey's procedure. 7-hydroxyheptaphylline 2 was obtained from 21 by treatment with DIBAL-H. A similar procedure was performed to prepare 7-methoxyheptaphylline 3 from aniline 14 and bromide 22 (Scheme 1).
Synthesis of Heptaphylline 1, 7-Hydroxyheptaphylline 2, 7-Methoxyheptaphylline 3, Mukoenine-B 4 (clausenatine-A), Euchrestine-A 5, and Isomurrayafoline-B 6.
Reaction between 2-hydroxycarbazole 11 with carbonate 28 furnished corresponding pyrano[3,2-a]carbazole 29, which was converted to mukoenine-B 4 in 21% (along with 50% of Z isomer). Euchrestine-A 5 and isomurrayafoline-B 6 was synthesized from compound 30, which was prepared previously by the group.9 Reduction of 30 using DIBAL-H in the presence of SiCl4 followed by removal of the silyl group provided euchrestine-A. On the other hand, reductive pyran ring opening of 30 followed by first methylation and then TIPS deprotection furnished isomurrayafoline-B (Scheme 1).
In another study, the group reported the synthesis of the naturally occurring 2,7-dioxygenated formylcarbazole alkaloids 7-methoxymukonal 31, 7-methoxy-O-methylmukonal 32, and murrayalines -A-C 33-35 (Scheme 2).10
Total Synthesis of 2,7-Dioxygenated Formylcarbazole Alkaloids.
The Buchwald-Hartwig coupling of the arylamine 36 and bromide 22 gave diaryl amine 37, which underwent palladium(II)-catalyzed oxidative cyclization to form carbazole 38 then 3-formyl-carbazole 39 after DDQ oxidation. Removal of the benzyl ether in 39 was accomplished by using zinc bromide and 2,6-dimethoxytoluene to generate 7-methoxymukonal 31. Similarly, diaryl amine 43 was prepared from aniline 40 and bromide 22. Oxidative cyclization of 43 catalyzed by palladium(II) acetate produced carbazole 44, which then was oxidized to 7-methoxy-O-methylmukonal 32 by DDQ (Scheme 2).
Coupling reaction between arylamine 40 and chloride 45 delivered diaryl amine 46, which cyclized to carbazole 47. DIBAL-reaction of the nitrile group in 47 afforded murrayaline-A 33. A similar route was applied to prepare carbazole derivative 50 from aniline 48 and chloride 45. Removal of the TIPS protecting group in 50 by TBAF provided murrayaline-B 34. For the synthesis of murrayaline-C 35, carbazole 54 was obtained from arylamine 52 and chloride 45 through a sequence of Buchwald-Hartwig amination and palladium(II)-catalyzed oxidative cyclization. The ester 54 was reduced to alcohol 55 before converting to aldehyde 56. Finally, cleavage of TIPS group in 56 by TBAF furnished murrayaline-C 35 (Scheme 7).
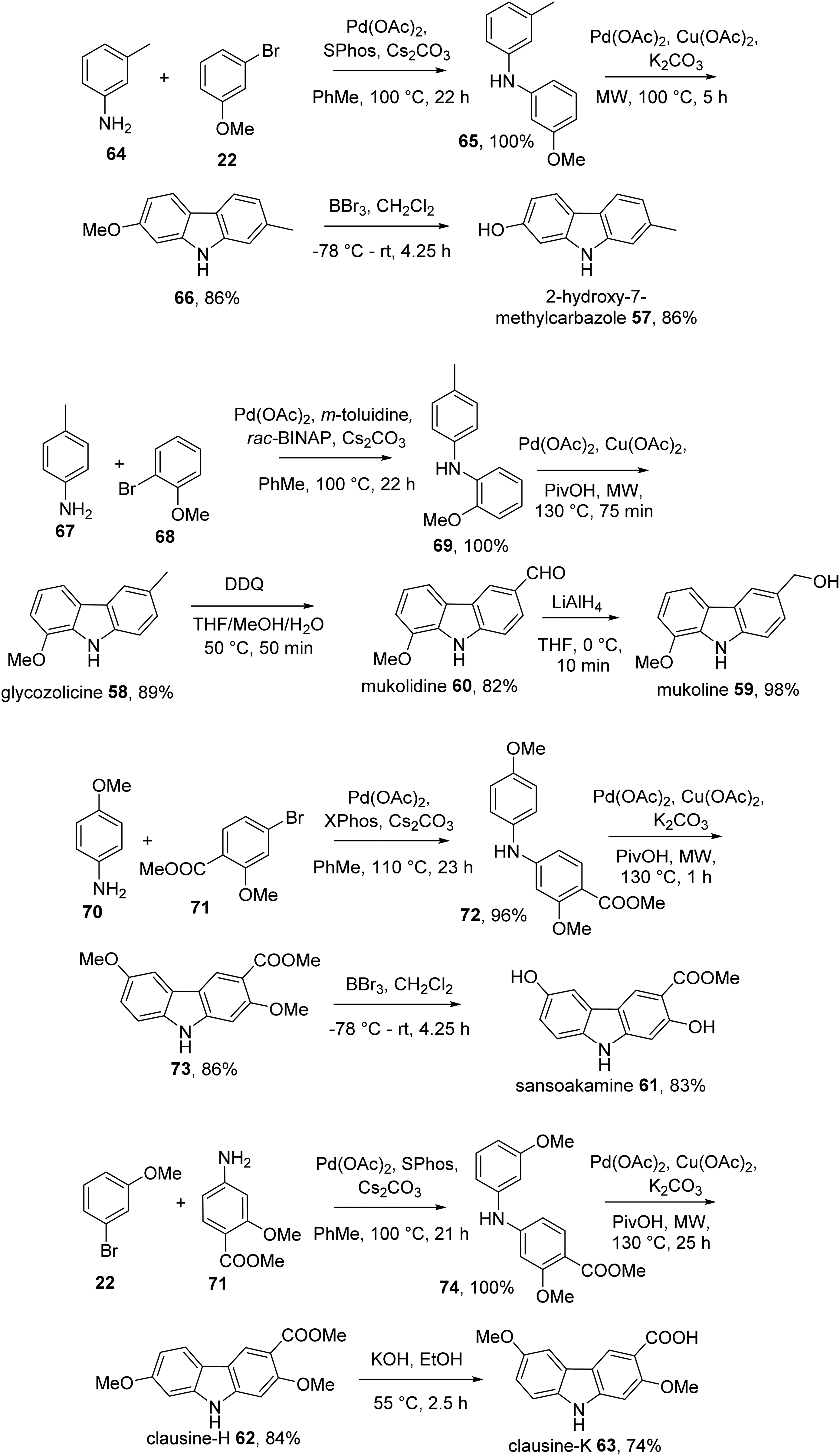
The group also successfully applied the palladium-catalyzed approach to the total synthesis of the eight carbazole alkaloids including 2-hydroxy-7-methylcarbazole 57, glycozolicine 58, mukoline 59, mukolidine 60, sansoakamine 61, clausine-H 62, and clausine-K (clauszoline-J, clausine-TY) 63.11
For the synthesis of 2-hydroxy-7-methylcarbazole 57, a Buchwald-Hartwig coupling reaction between m-bromoanisole 22 and m-toluidine 64 using catalytic amounts of palladium(II) acetate and SPhos as ligand delivered diarylamine 65, which underwent oxidative cyclization to form 2-methoxy-7-methylcarbazole 66. BBr3-catalyzed cleavage of methyl ether in 66 gave 57 in 87% yield. Similarly, coupling reaction between m-bromoanisole 68 and p-toluidine 67 generated diarylamine 69, which was converted to glycozolicine 58 by an oxidative cyclization process. Oxidation of the methyl group in 58 by DDQ gave mukolidine 60, which was reduced to mukoline 59 by LiAlH4 (Scheme 3).
Buchwald-Hartwig coupling of methyl 4-bromo-2-methoxybenzoate 71 with aniline 70 resulted in diarylamine 72, which cyclized to carbazole 73 using Pd(II) catalyst. Cleavage of two methyl ethers in 73 afforded sansoakamine 61. For the synthesis of clausine-H 62 and clausine-K 63, coupling reaction between aniline 71 and bromide 22 yielded diarylamine 74, which was transformed into clausine-H 62. Finally, cleavage of the methyl ester of 62 produced clausine-K 63 (Scheme 3).
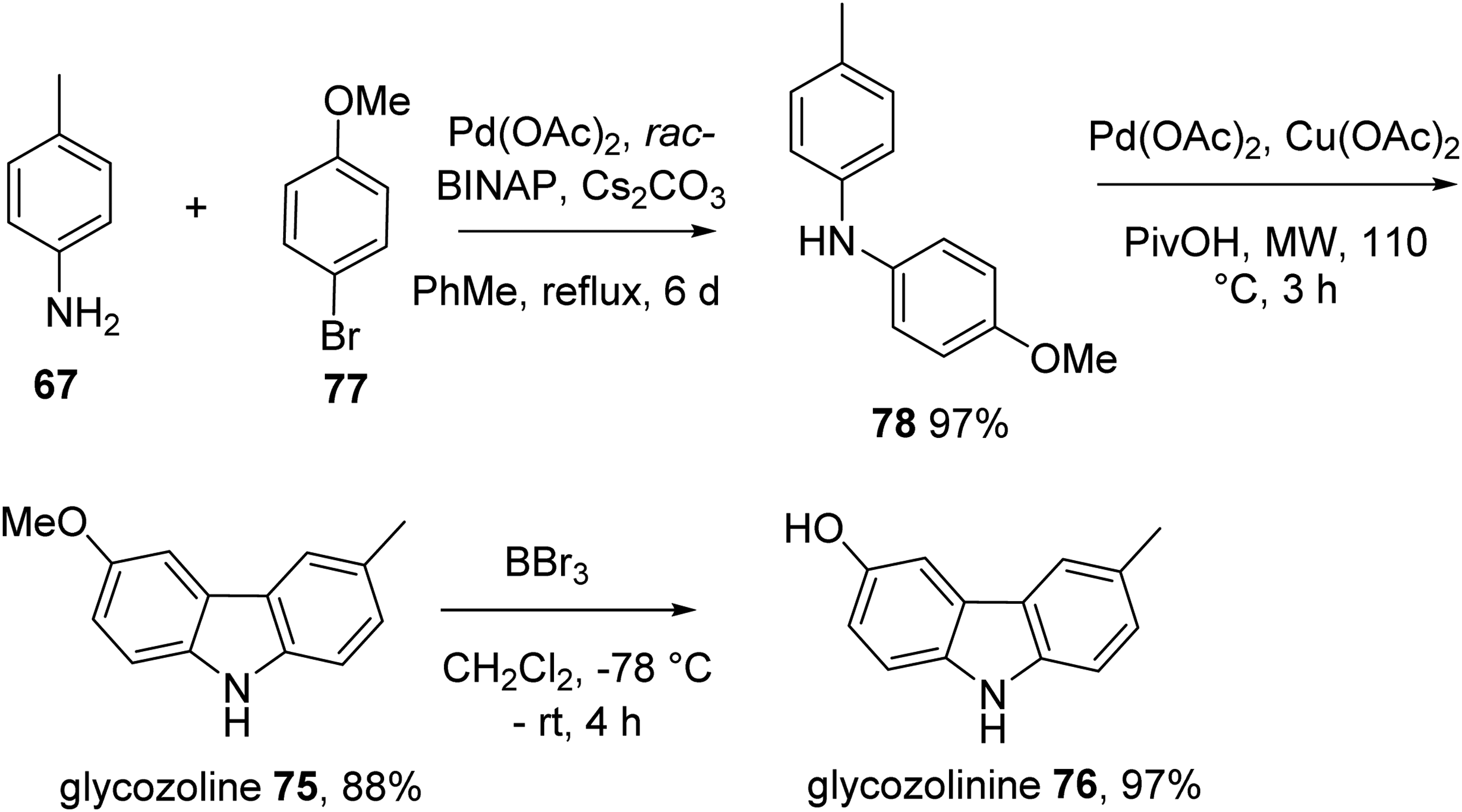
In 2015, the group accomplished the total synthesis of glycozoline 75 and glycozolinine 76.12 Buchwald-Hartwig coupling of p-bromoanisole 77 and p-toluidine 67 catalyzed by palladium(II) acetate and rac-BINAP produced diarylamine 78, which underwent oxidative cyclization to furnish glycozoline 75. BBr3-catalyzed cleavage of methyl ether in glycozoline gave glycozolinine 75 (Scheme 4). All reaction were performed with excellent yields.
Total Synthesis of Glycozoline 75 and Glycozolinine 76.
In 2016, the group accomplished the total synthesis of murrayaquinones -B-E 79-82 and murrayafoline-B 83.13 Copper(I)-catalyzed Ullmann-type coupling of m-iodoanisole 84 and aniline 85 gave diarylamine 86, which was protected as tosylate 87. Pd-catalyzed oxidative cyclization of 87 provided carbazole 88, which was converted to 89 by treatment with HBr. Reaction of 89 with 1,1-dimethylpropargyl methyl carbonate 12 catalyzed by copper(II) chloride and DBU afforded propargyloxycarbazole 90, which underwent thermally-induced rearrangement to produce pyrano[3,2-a]carbazole 91. This pyranocarrbazole was transformed into 93 by a sequence of DIBAL-H reduction and methylation. Removal of the Ts protecting group in 93 by LiAlH4 furnished murrayafoline-B 83. Treatment of murrayafoline-B with Frémy's salt delivered murrayaquinone-B 79. Murrayaquinone-E 82 was obtained from 93 by removal of Ts group by LiAlH4 followed by treatment with Frémy's salt (Scheme 5).
Total Synthesis of Murrayaquinones -B-E 79-82 and Murrayafoline-B 83.
Copper(II)-catalyzed reaction of 2-hydroxycarbazole 89 with carbonate 28 and DBU yielded ether 95, which was converted to 96 by Lindlar reduction. Thermally induced Claisen rearrangement of 96 gave compound 97 as a mixture of E and Z isomers. Methylation of 97 followed by Ts removal using LiAlH4 provided mixture 99. Oxidation of 99 with Frémy's salt, and separation of the isomers resulted in murrayaquinone-C 80. Similarly, murrayaquinone-D 81 was prepared from 97 in two steps (Scheme 5).
In the same year, the group described the total syntheses of glycozolidol 101 and carbalexin-C 102.14 Buchwald-Hartwig coupling of bromide 103 and aniline 40 catalyzed by Pd(OAc)2 in the presence of DavePhos as ligand produced diarylamine 104, which underwent palladium(II)-catalyzed oxidative cyclization to give carbazole 105. Cleavage of TBDPS group of 105 by TBAF furnished glycozolidol 101. An analogous procedure was applied to prepare carbalexin-C 102 from bromide 77 and aniline 106 (Scheme 6).
Total Synthesis of Glycozolidol 101 and Carbalexin-C 102.
In another report, the group demonstrated the total synthesis of mukonine 109, murrayafoline-A 110, and clausine-L 111.15 Pd(II)-catalyzed coupling reaction between aniline 112 and bromobenzene 113 yielded diarylamine 114, which was transformed into mukonine 109 by palladium(II)-catalyzed oxidative cyclization. Reduction off the ester group in 109 using LiAlH4 then furnished murrayafoline-A 110. The same proedure was performed to prepare clausine-L 111 from aniline 71 and iodide 8 (Scheme 7).
Total Synthesis of Mukonine 109, Murrayafoline-A 110, and Clausine-L 111.
The group also accomplished the total synthesis of glycoborine 116, carbalexin-A 117, glybomine-A 118, and glybomine-B 119.16 Buchwald-Hartwig coupling of bromophenyl pivalate 120 and p-toluidin 67 afforded diarylamine 121, which underwent oxidative cyclization to form carbazole 122. This carbazole was converted to glycoborine 116 by a sequence of ester reduction by LiAlH4 and methylation. Similarly, carbazole 128 was obtained from bromide 148 and aniline 120 through a sequence of Buchwald-Hartwig coupling, oxidative cyclization, ester reduction, and methylation. Removal of the TIPS protecting group in 128 furnished carbalexin-A 117, which could be further methylated to glybomine-A 118. An analogous route was performed to prepare glybomine-B 119 from bromide 120 and aniline 40 (Scheme 8).
Total Synthesis of Glycoborine 116, Carbalexin-A 117, Glybomine-A 118, and Glybomine-B 119.
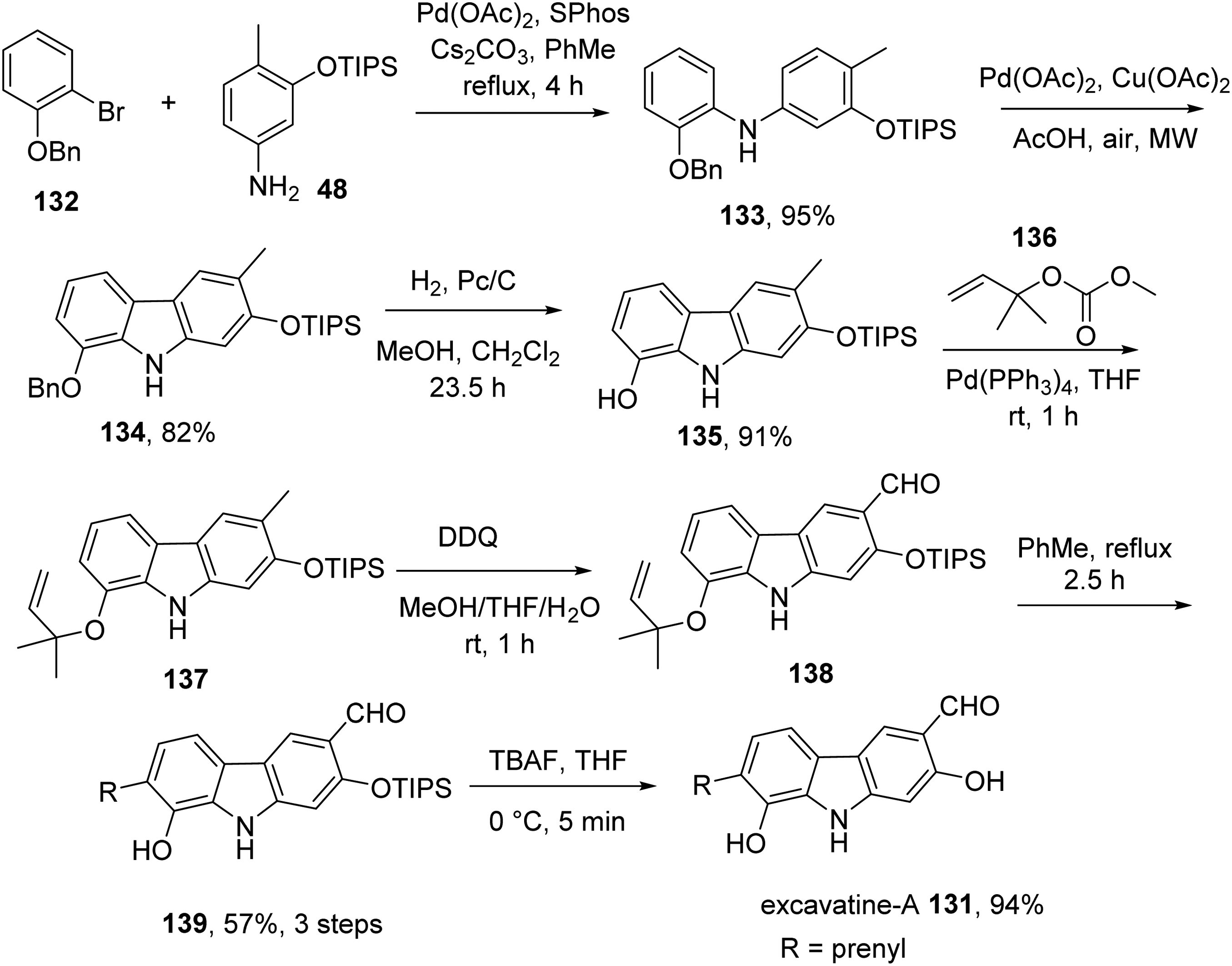
In 2017, the group achieved total synthesis of excavatine-A 131 in 7 steps and 38% overall yield.17 Palladium-catalyzed coupling of bromide 132 and aniline 124 using SPhos as ligand and cesium carbonate as base delivered diarylamine 133. Microwave irradiation of a mixture of 133, pivalic acid, copper(II) acetate, and palladium(II) acetate resulted in carbazole 134, which was converted to 135 by hydrogenolytic cleavage of the benzyl ether. Reaction between 135 and 1,1- dimethylallyl methyl carbonate 136 gave ether 137, which was oxidized to aldehyde 138 using DDQ. Claisen rearrangement of 138 under thermal conditions yielded prenylated carbazole 139, which was transformed into excavatine-A 131 by treatment with TBAF to remove the TIPS group (Scheme 9).
Total Synthesis of Excavatine-A 131.
In 2020, the group reported the total synthesis of clausenal 140.18 Buchwald-Hartwig coupling of aniline 112 and bromide 68 produced diarylamine 141, which underwent palladium(II)-catalyzed oxidative cyclization to generate carbazole 142. Reduction of thester group in 142 yielded primary alcohol 143, which was reduced to clausenal 140 by using MnO2 (Scheme 10). Clausenal was achived in four steps in 73% overall yield.
Total Synthesis of Clausenal 140.
Synthesis of Simple Natural Carbazole Alkaloids by Other Groups
Ma et al demonstrated the total gram-scale synthesis of natural carbazole alkaloids including 7-methoxy-O-methylmukonal 32, 7-methoxymukonal 31, clausine-O 144, clausine-K 63, clausine-H 62, and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate (proposed structure of clausine-TY) 145 from indole-2-carbaldehyde 146.19
Reaction between indole-2-carbaldehyde 146 and 1-methoxypropa-1,2-diene 147 yielded allenol 148, which was transformed into carbazole 149via an efficient Au-catalyzed cyclization. Bromination of 149 using NBS afforded 150 and NH carbazole 151 after debenzylation using molecular oxygen under basic conditions. Treatment of 151 with KH in THF followed by reaction with BuLi in DMF produced 7-methoxy-O-methylmukonal 32. Cleavage of two ether groups in 32 by treatment with BBr3 then furnished clausine-O 144. Treatment of 32 with AlCl3 led to the selective removal of methyl ether at C3 and 7-methoxymukonal 31 was produced in 94% yield. Deprotonation of 151 with KH in THF followed by treatment with BuLi, CO2 in THF afforded clausine-K 63, which could be esterified to clausine-H 62. Treatment of clausine-H 62 with AlCl3 gave proposed structure of clausine-TY 145 (Scheme 11). However, spectroscopic data of 145 did not match with those data reported by Taufiq-Yap et al20 and the authors suggested that the structure of clausine-TY 145 should be revised. Actually, in a total synthesis study, Knöller et at. revised structure of clausine-TY as clausine-K.11
Total Synthesis of 7-Methoxy-O-Methylmukonal 32, 7-Methoxymukonal 31, Clausine-O 144, Clausine-K 63, Clausine-H 62, and Methyl 2-Hydroxy-7-Methoxy-9H-Carbazole-3-Carboxylate 145 by Ma et al.
Markad and Argade demonstrated an efficient route for the formal synthesis of carbazomycin A 152, carbazomycin B 153, hyellazole 154, and chlorohyellazole 155.21 Reaction between Boc-protected 3-formylindole 156 and an in situ generated Wittig reagent from dimethyl maleate 157 gave 158, which was methylated with MeI, LiHMDS to form 159. Regioselective coupling reaction of N,O-dimethylhydroxylamine hydrochloride with 159 furnished 160. Chemoselective nucleophilic additions of methylmagnesium bromide 161 and phenylmagnesium bromide 162 to Weinreb amide 160 provided ketone 163 and 164, respectively. p-TSA-catalyzed intramolecular cyclization of these ketones delivered the corresponding carbazole esters 165 and 166. Reduction of these esters afforded the corresponding primary alcohol 167 and 168, which were oxidized to aldehydes 169 and 170. The Baeyer-Villiger oxidation of these aldehydes resulting the corresponding formate esters and then phenolic compounds 171 and 172 after hydrolysis. Methylation of these phenolics by MeI, K2CO3 afforded carbazoles 173 and hyellazole 154, two precusors of carbazomycin A 152, carbazomycin B 153, and chlorohyellazole 154 (Scheme 12).22
Total Synthesis of Carbazomycin A 152, Carbazomycin B 153, Hyellazole 154, and Chlorohyellazole 155 by Markad and Argade.
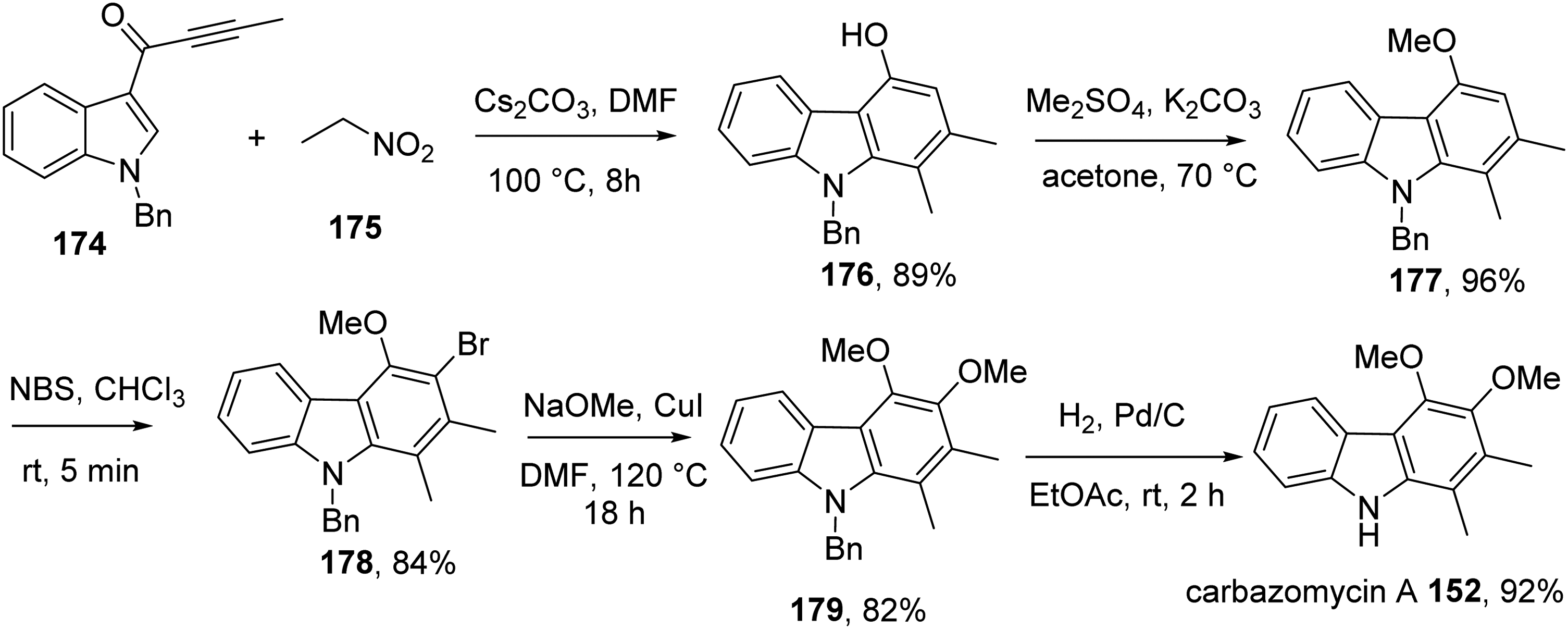
Singh et al developed a short and efficient strategy for the synthesis of carbazomycin A 152.23 Initially, the one-pot domino benzannulation reaction between indole-3-ynone 174 and nitroethane 175 catalyzed by Cs2CO3 furnished carbazole 176 in excellent yield. Methylation of 176 gave 177, which underwent bromination with NBS to afford bromide 178. This bromide was converted to 179, which was transformed to carbazomycin A 152 by hydrolytic debenzylation (Scheme 13). The synthesis gave carbazomycin A 152 in 54% overall yield over five steps.
Total Synthesis of Carbazomycin A 152 by Singh et al.
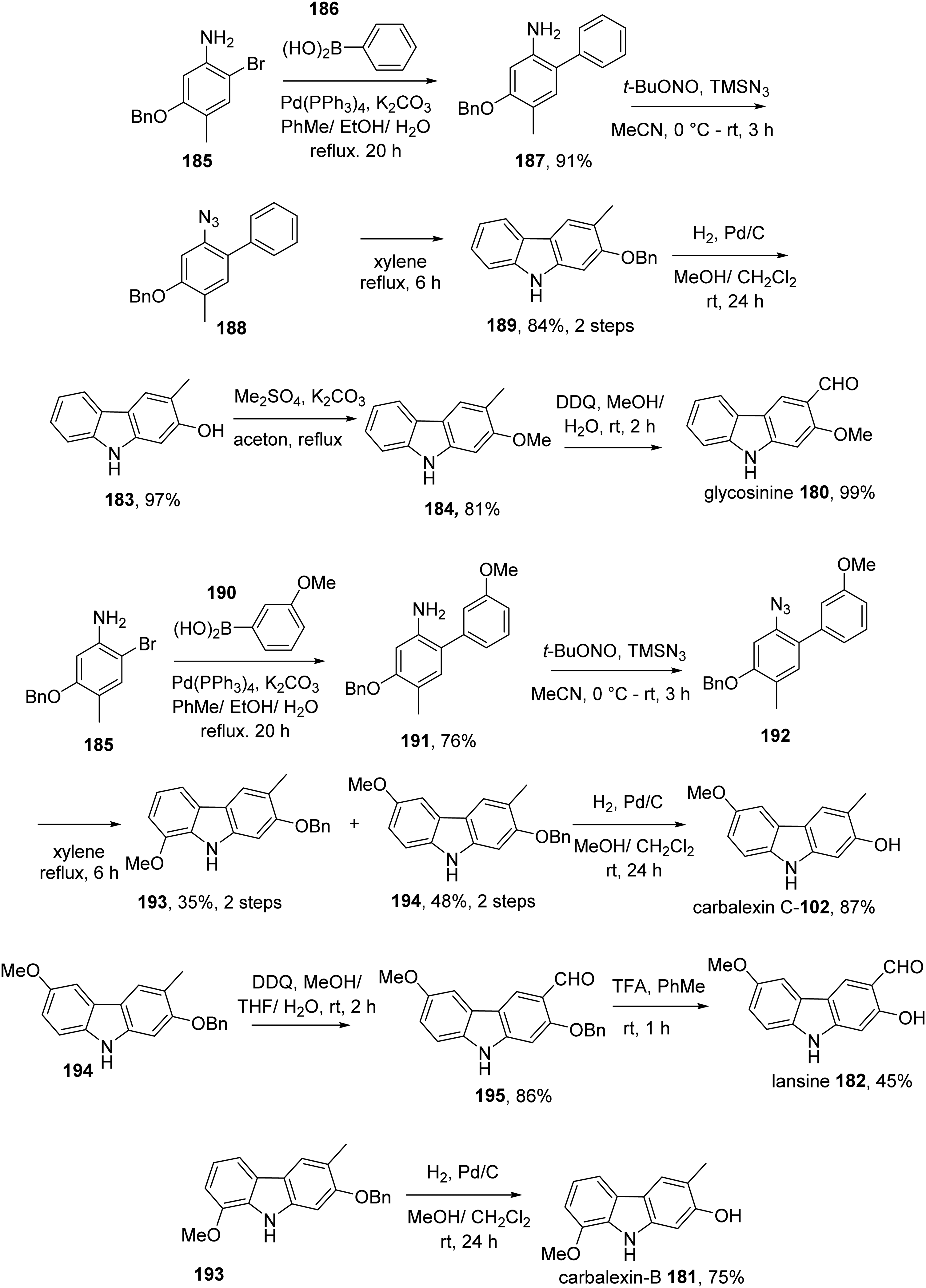
Polley et al described the total synthesis of glycosinine 180, carbalexin-B 181, lansine 182, carbalexin-C 102, 2-hydroxy-3-methyl carbazole 183, and 2-methoxy-3-methyl carbazole 184.22 Suzuki coupling reaction of aryl bromide 185 with phenyl boronic acid 186 in the presence of catalytic Pd(PPh3)4 in EtOH/toluene gave biaryl amine 187. This aniline was converted to the corresponding azide 188via diazotization with tert-butyl nitrite and trimethylsilyl azide. Next, the intramolecular C-N coupling of this azide was performed at refluxing xylene to afford carbazole 189. Debenzylation of 189 produced 2-hydroxy-3-methylcarbazole 183, which was converted to 184 by methylation. Oxidation of 184 using DDQ resulted in glycosinine 180. A similar route was applied for the synthesis of carbalexin-C 181 from bromide 185 and borononic acid 190. Oxidation of 190 by DDQ delivered aldehyde 191, which was transformed into lansine 182 TFA-promoted debenzylation. Carbalexin-B 224 was also obtained from 233 by benzyl removal (Scheme 14).
Total Synthesis of Glycosinine 180, Carbalexin-B 181, Lansine 182, Carbalexin-C 102, 2-Hydroxy-3-Methyl Carbazole 183, and 2-Methoxy-3-Methyl Carbazole 184 by Polley et al.
Roy and Mal performed a short total synthesis of ekeberginine 196 based on anionic [4 + 2] annulation of N-methoxymethyl- furoindolone 197 with dimethyl maleate.24 Alkylation of 197 by prenyl bromide 198 provided prenylfuroindolone 199, which underwent annulation with dimethyl maleate and LDA to generate adduct 200. The reaction of 200 with sodium chloride in dimethyl sulfoxide at reflux resulted in decarboxylated carbazole, which was converted to 201 by methylation. Finally, ekeberginine 196 was achieved from 201 by DIBAL-H reduction and MnO2 oxidation (Scheme 15). Ekeberginine 196 was prepared in 42% overall yield in four steps.
Total Synthesis of Ekeberginine 196 by Roy and Mal.
Later, these authors applied the abovementioned approach for the total synthesis of murrayafoline A 202 and clausamine E 203.25 Annulation reaction of Boc-protected furoindolone 204 with methyl crotonate 205 afforded carbazole 206, which underwent methylation with MeI to form 207. The ester 207 was then converted to the corresponding acid, which was subjected to decarboxylation using Cu2O, TMEDA in NMP to furnished murrayafoline A 202. Boc protection of 202 gave 208, which was brominated to 209 by using NBS. The methyl group in 209 was then oxidized to aldehyde 210 by MnO2 and acid 211 by NaClO2. Methylation of this acid delivered ester 212, which underwent Heck-type reaction with 2-methyl-but-3-en-2-ol delivered clausamine E 203. Clausamine E was achieved in 11 steps from readily accessible furoindolone 204 (Scheme 16).
Total Synthesis of Murrayafoline A 202 and Clausamine E 203 by Roy and Mal.
Humne et al described the total synthesis of glycozoline 75 and murrayafoline A 202 in short routes.26 The reaction between phenylhydrazine 213 and 4-methyl-cyclohexanone 214 provided tetrahydrocarbazole 215, which was transformed into glycozoline 75 in excellent yield by treatment with iodine in DMSO. For the synthesis of murrayafoline A 202, tetrahydrocarbazole 217, which was obtained from phenylhydrazine 216 and 4-methyl-cyclohexanone 214, was oxidized to ketone 218 by H5IO6. Oxidation of this ketone afforded carbazole 219, which was converted to murrayafoline A 202 by methylation reaction using CH2N2 (Scheme 17).
Total Synthesis of Glycozoline 75 and Murrayafoline A 202 by Humne et al.
In an analogous study, Naykode et al demonstrated an efficient approach to successfully accomplish a collective total synthesis of carbazole alkaloids including clauszoline-K 220, 3-formyl-7-hydroxycarbazole 221, glycozolicine 58, clausine-C 222, clausine-M 223, clausine-N 224, clauraila A 225, and clausenal 140.27 The reaction between phenylhydrazine 226 and 4-methyl-cyclohexanone 214 provided tetrahydrocarbazole 227, which was transformed into carbazole 228 by heating with iodine in DMSO. Compound 229 then was obtained from 228 by reaction with NaOMe, CuI. Oxidation of the methyl group in 229 furnished clauszoline K 220, which was converted to 3-formyl-7-hydroxycarbazole 221 by methyl ether cleavage using BBr3. A similar procedure was applied to prepare clausine C 222 from phenylhydrazine 226 and cyclohexanone 230. Treatment of clausine C 222 with NaOH in MeOH gave clausine N 224, while BBr3-promoted removal of the methyl ether of 222 delivered clausine M 223. Following the same route, glycozolicine 58 was achieved from phenylhydrazine 233 and 4-methyl-cyclohexanone 214 (Scheme 18).
Total Synthesis of Clauszoline-K 220, 3-Formyl-7-Hydroxycarbazole 221, Glycozolicine 58, Clausine-C 222, Clausine-M 223, Clausine-N 224, Clauraila A 225, and Clausenal 140 by Naykode et al.
For the synthesis of clauraila A 225, the tetrahydrocarbazole 227 was oxidized to ketone 236 by H5IO6 before converting to carbazole 237. Methylation of 237 afforded 238, which was converted to 239 by subjecting to NaOMe, CuI. Finally, oxidation of 239 by DDQ furnished clauraila A. Following the same route, clausenal 140 was obtained from the tetrahydrocarbazole 252 in five steps (Scheme 18). In most cases, reaction was performed in good to excellent yield.
Dhara et al demonstrated the total synthesis of girinimbilol 244 and heptaphylline 1 starting from isatin derivative 245.28 Benzyl protection of 245, followed by a Grignard reaction with allylmagnesium bromide 247, gave the desired diallyl-3-oxindole 248. Ring closing metathesis of 248 afforded spirocyclic 3-oxindole 249, which underwent one-pot rearrangement-aromatization to provide carbazole 250. These two steps are key feature of the synthesis. Hydrogenolysis of the benzyl protecting groups in 250 resulted in 2-hydroxy-3-methylcarbazole 183, which was converted to girinimbilol 244 by prenylation. Oxidation of 244 then furnished heptaphylline 1 (Scheme 19).
Total Synthesis of Girinimbilol 244 and Heptaphylline 1 by Dhara et al.
Banerjee et al synthesized hyellazole 154 and 6-chlorohyellazole 155 based on benzannulation of 2-alkenylindoles and methoxyacetaldehyde 251.29 The reaction between 2-alkenylindole 252 and methoxyacetaldehyde 251 produced hyellazole 154, while the use of 2-alkenylindole 253 furnished 6-chlorohyellazole 155 (Scheme 20).
Total Synthesis of Hyellazole 154 and 6-Chlorohyellazole 155 by Banerjee et al.
Chakraborty and Saha introduced a route for the total synthesis of hyellazole 154.30 Sandmeyer reaction of aniline 254 afforded bromide 255, which was reduced to 256 using Fe/HCl. 3-bromo-2-methylphenol 257 then was obtained from 256 through diazotization. Coupling reaction of 257 with diazonium salt 258 provided 4-amino-3- bromo-2-methylphenol 260via azo intermediate 259. Amine hydrochloride 263 was then achieved from 260 by a sequence of N-acetylation, methylation, and hydrolysis of acetamide derivative. Amine hydrochloride 263 was converted to the corresponding diazonium salt 264, which then underwent Japp-Klingemann coupling with 2-formylcyclohexanone 265 generated substituted phenylhydrazonocyclohexanone 266. Fischer indole cyclization of 266 afforded 1-ketotetrahydrocarbazole 267, which was transformed into carbazole 268 by a sequence of Wolff-Kishner reduction and aromatization. Finally, Suzuki cross-coupling of 268 with phenyl boronic acid furnished hyellazole 154 (Scheme 21).
Total Synthesis of Hyellazole 154 by Chakraborty and Saha.
In 2017, Alimi et al accomplished the total synthesis of clausenawalline D 269 based on the photolytic reaction of N-arylbenzotriazole.31 Reaction between aromatic azide 270 with the benzyne precursor 271 provided N-arylbenzotriazole 272, which underwent hydrolytic removal of Bn group to afford 273. Irradiation of 273 for 24 h in acetonitrile in a Rayonnet® photochemical reactor, equipped with 8 UV lamps (254 nm) furnished clausenawalline D 269 (Scheme 22).
Total Synthesis of Clausenawalline D 269 by Alimi et al.
Qiu et al developed a simple and efficient method for the synthesis of polysubstituted 2-oxygenated carbazoles by [IPrAuCl]/AgSbF6-catalyzed cyclization of the readily available 4-benzoxyl-1-(indol-2-yl)-2-alkynols and applied this method to prepare karapinchamine A 274.32 Alkylation of ethyl 5-methyl-1H-indole-2-carboxylate 275 by (2E)-geranyl bromide 276 gave indole 277, which was reduced to primary alcohol 278 using LiAlH4. This alcohol was oxidized to aldehyde 279, which underwent nucleophilic addition with organometallic compound 280 to provide alcohol 281. [IPrAuCl]/AgSbF6-catalyzed cyclization of this 4-benzoxyl-1-(indol-2-yl)-2-alkynol furnished carbazole 282. Removal Bz group from 282 by treatment with K2CO3 delivered karapinchamine A 274 (Scheme 23).
Total Synthesis of Karapinchamine A 274 by Qiu et al.
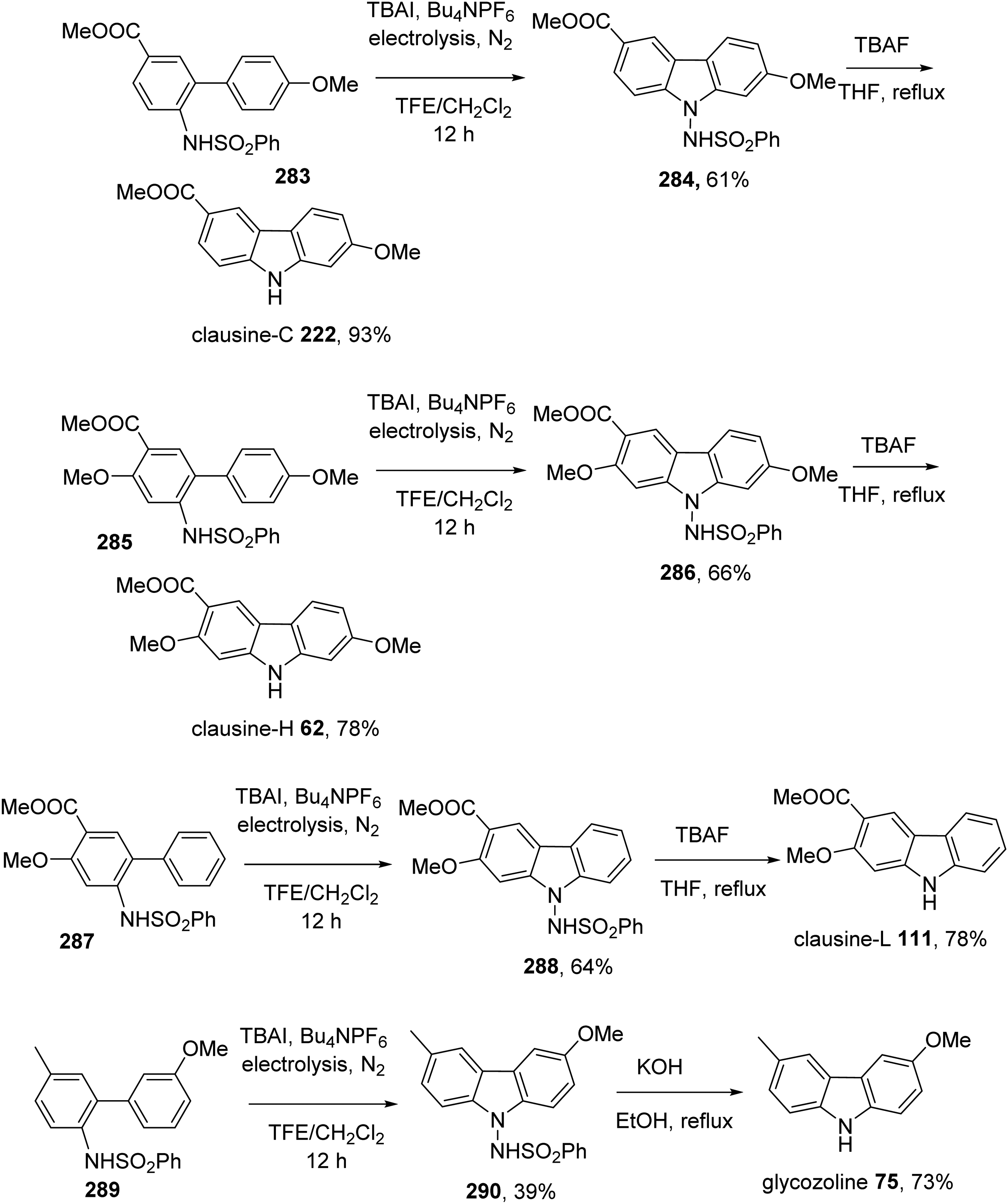
Zhang et al introduced an efficient electrochemical C-H bond dehydrogenative amination protocol for the construction of carbazoles from 2-amidobiaryls. The approach was successfully applied to prepare clausine-C 222, clausine-H 62, clausine-L 111, and glycozoline 75.33 Cyclization of 283 under electrolytic condition gave carbazole 284. Treatment of 284 with TBAF then afforded clausine C 222. Similarly, clausine H 62, clausine L 111, and glycozoline 75 were obtained from 2-amidobiaryls 285, 287, and 289, respectively (Scheme 24).
Total Synthesis of Clausine-C 222, Clausine-H 62, Clausine-L 111, and Glycozoline 75 by Zhang et al.
Total Synthesis of Glycozoline 75, Glycozolicine 58, and Murrayafoline A 202 by Chakraborty et al.
Chakraborty et al demonstrated an efficient approach toward the synthesis of carbazoles from 1-keto-1,2,3,4-tetrahydrocarbazoles by a sequence of tosylhydrazones formation and one-pot tandem reduction-oxidation of resulting tosylhydrazones by using a combination of NaBH4 and Pd/C on MgSO4.7H2O support under microwave irradiation. Glycozoline 75 and glycozolicine 58 were synthesized in high yields. The approach was also utilized for the total synthesis of murrayafoline A 202.34 For the which synthesis of murrayafoline A 202, aniline 348 was obtained from phenol 345 through a sequence of nitration, methylation, and nitro reduction. This aniline was converted to the corresponding diazonium salt 349, which then underwent Japp-Klingemann coupling with 2-formylcyclohexanone 314 afforded phenylhydrazonocyclohexanone 350. Fisher indole synthesis using 350 under acidic conditions gave keto-1,2,3,4-tetrahydrocarbazole 351, which was transformed into murrayafoline A 22 by the described procedure (Scheme 32).
Elumalai et al completed the total synthesis of carbazole alkaloid carbazomycin G 303 in an overall yield of 8.3%.35 The synthesis employed 304 as the starting material. Oxidation of 304 with hydrogen peroxide afforded phenol 305, which was acetylated to acetate 306. Nitration of 306 delivered nitro 307 in high yield, which was converted to 308 by acid treatment. Chlorination of 308 with N,N′-dichloro-5,5-dimethylhydantoin 309 produced 310, which was transformed into 311 by Suzuki cross-coupling phenylboronic acid 186. Reduction of 311 by In/HCl provided aniline 312, which was then protected as acetamide 313. Cyclization of 313 using Pd(OAc)2 and Imes.HCl as a catalyst system delivered carbazole 314. Removal of the acetyl group in 314 generated NH-carbazole 315, which was oxidized to 316 by HNO3. Finally, regioselective methylation of 316 using MeLi furnished carbazomycin G 303 (Scheme 26).
Total Synthesis of Carbazomycin G 303 by Elumalai et al.
Kumar et al described a simple and efficient route for the total synthesis of murrayafoline A 202, clausenine 317, clauraila A 225, and clausenal 140.36N-arylation of isatin 318 with phenylboronic acid 319 catalyzed by CuOTf and 1,10-phenanthroline gave 320 in excellent yield. Cleavage of the N-arylated benzoxazolone 320 moiety under basic conditions (NaOH, EtOH/H2O, 60 °C) followed by O-methylation with MeI/ K2CO3 afforded diarylamine 321, which was transformed into murrayafoline A 202 by treatment with Pd(OAc)2/K2CO3 in pivalic acid at elevated temperature. The same route was performed to prepare clausenine 317, carbazoles 239 and 243 from isatin 322 and boronic acids 323, 190, and 328, respectively. Then oxidation of carbazoles 229 and 243 furnished clauraila A 225 and clausenal 140, respectively (Scheme 27).
Total Synthesis of Murrayafoline A 202, Clausenine 317, Clauraila A 225, and Clausenal 140 by Kumar et al.
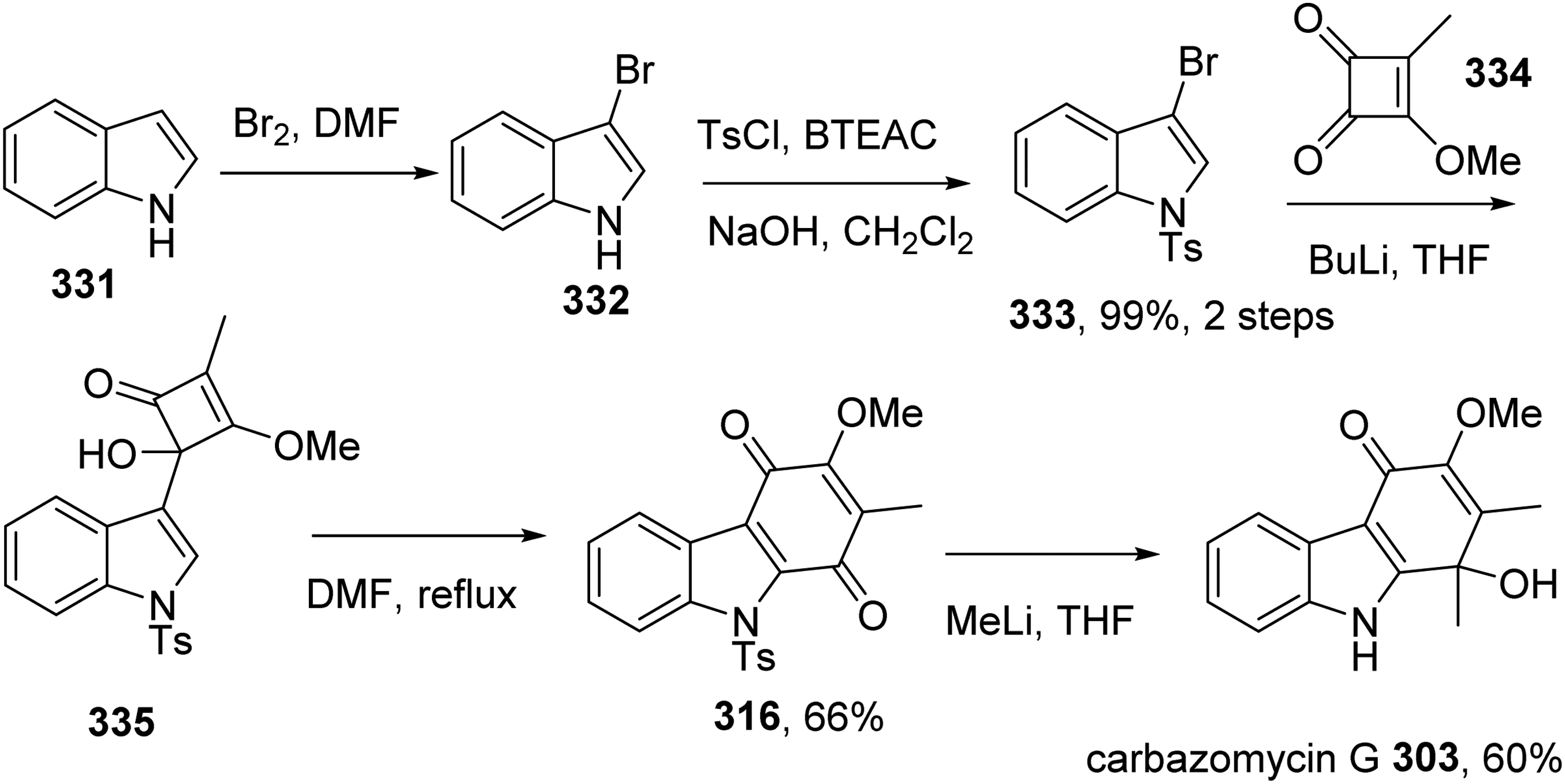
An et al introduced the total synthesis of carbazomycin G 303 from indole in six steps and 26% overall yield.37 Bromination of indole 331 followed by tosyl protection afforded 333. Treatment of 333 with BuLi followed by reaction with cyclobutenedione 334 furnished 335, which was transformed into carbazolequinone 316. Methylation of 316 with MeLi then provided carbazomycin G 303 (Scheme 28).
Total Synthesis of Carbazomycin G 303 by An et al.
Liu et al accomplished the total synthesis of clausenapin 336 and indizoline 337 from indole- 3-carbaldehyde 338.38 Stobbe condensation of indole- 3-carbaldehyde 338 with dimethyl succinate 339 promoted by sodium hydride afforded succinic monoester 340, which underwent TFAA-mediated intramolecular acylation to give carbazole 341. Allylation of 341 generated allyl ether 342, which was converted to 343 by a sequence of Claisen rearrangement and methylation. Redcution of 343 by LiAlH4 resulted in 344. Finally, cross diene metathesis of 344 with 2,3-dimethyl-2-butene 345 furnished clausenapin 336 in excellent yield. On the other hand, propargylation of 341 with 1,1- dimethylpropargyl trifluoroacetate 346 produced 347, which then underwent hydrogenation under Lindlar's cataalysis to form 348. Methylation of 348 delivered 349, which was transformed into indizoline 337 by a sequence of LiAlH4 reductiom and DMP oxidation (Scheme 29).
Total Synthesis of Clausenapin 336 and Indizoline 337.
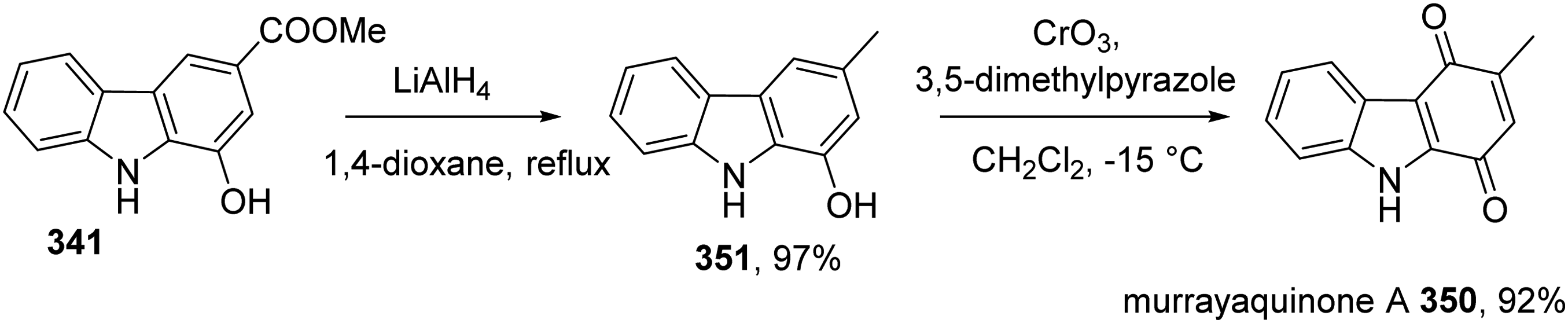
Ji et al prepared murrayaquinone A 350 in two steps from carbazole 341,39 which was prepared following literature report.38 Reduction of ester 341 by LiAlH4 in dioxane resulted in 351. This compound was oxidized by chromium trioxide to afford murrayaquinone A 350 (Scheme 30).
Synthesis of Murrayaquinone A 350 by Ji et al.
Hernández-Benitez et al demonstrated the total synthesis of series of carbazole alkaloids including clauraila A 225, clausenal 140, clausine-P 352, and 7-methoxy-O-methylmukonal 32.40 Reaction between aniline 353 and cyclohexan-1,3-dione 354 gave enamine 355, which underwent aromatization followed by methylation to give diarylamine 327. Pd-catalyzed oxidative cyclization of 327 afforded carbazole 229, which was oxidized to clauraila A 225 by DDQ. Similarly, clausenal 140 was obtained following the same procedure from aniline 353 and cyclohexan-1,2-dione 356. For the synthesis of clausine P 4352, aniline 40 and cyclohexan-1,2-dione 356 were employed, while the use of aniline 40 and cyclohexan-1,3-dione 354 furnished 7-methoxy-O-methylmukonal 32 (Scheme 31).
Total Synthesis of Clauraila A 225, Clausenal 140, Clausine-P 352, and 7-Methoxy-O-Methylmukonal 32 by Hernández-Benitez et al.
Rasheed et al accomplished the total synthesis of clausine-C 222, clausine-L 111, clausine-H 62, glycozoline 75, glycozolidine 361, glycozolidal 362, and sansoakamine 61 based on copper promoted N-arylation of methyl 4-amino-3-iodobenzoate with boronic acids.41N-arylation of methyl 4-amino-3-iodobenzoate 363 by boronic acid 190 catalyzed by Cu(OAc)2 and 2,6-lutidine in toluene under air and n-decanoic acid gave 2-iodo diarylamine 364. Intramolecular C-H arylation of 364 using Pd(OAc)2 (10 mol%) as catalyst, K2CO3 as base and DMSO as solvent at elevated temperature furnished clausine C 222. Similarly, clausine H 62 was synthesized from iodoaniline 365 and boronic acid 190. For the synthesis of clausine L 111, iodoaniline 365 and boronic acid 186 were employed. Following the same route, carbazoles 417 and 419 were achieved from anilines 437 and 435 with boronic acid 387, respectively. Reduction of 437 and 435 by LiAlH4 then furnished glycozolidine 433 and glycozoline 117, respectively. Treatment of 441 with BBr3 afforded sansoakamine 103, while reduction of 417 by DIBAL-H provided glycozolidal 434 (Scheme 32).
Total Synthesis of Clausine-C 222, Clausine-L 111, Clausine-H 62, Glycozoline 75, Glycozolidine 361, Glycozolidal 362, and Sansoakamine 61 by Rasheed et al.
Kaliyaperumal et al completed the total synthesis of murrayaquinone A 350 in high yield from aldehyde 373.42 Iodination of this aldehyde produced 374, which underwent reductive amination with aniline 375 to afford diarylamine 376. Reduction of aldehyde 376 by TMSCl and TES gave 377, which was converted quinone 378 by CAN Oxidation. Murrayaquinone A 350 was furnished from 378 by intramolecular C–X/C–H cross coupling reaction. Alternatively, amination of aniline with iodide 445 delivered diarylamine 450, which underwent Pd-catalyzed oxidative cyclization to form carbazole 451. Reduction of this carbazole by TMSCl and TES generated 452, which was transformed into murrayaquinone A 408 by treatment with BBr2 in CH2Cl2 (Scheme 33).
Total Synthesis of Murrayaquinone A 350 by Kaliyaperumal et al.
Goo and Woo demonstrated highly efficient synthetic methods for carbazoles synthesis from boronic acids and ortho-bromonitrobenzenes.43 A sequence of C-C/C-N bond formations via Suzuki cross-coupling and reductive Cadogan cyclization occurred in one pot. The procedure was applied to prepare glycozoline 75, glycozolicine 58, and glycozolidine 361. Reaction between o-bromonitrobenzene 382 and boronic acid 230 gave glicozoline 75 and glycozolizine 58, while reaction between o-bromonitrobenzene 383 and boronic acid 384 furnished glycozolidine 361 (Scheme 34).
Synthesis of Glycozoline 75, Glycozolicine 58, and Glycozolidine 361by Goo and Woo.
Norcott and McErlean achieved the total synthesis of murrayaquinones- B-E 79-82 and königinequinone A 385.44 Reaction between toluhydroquinone 386 and m-anisidine 387 in aqueous hydrogen peroxide and a catalytic amount of iodine provided 388. Treatment of 388 with palladium acetate under refluxing acetic acid afforded königinequinone A 385, which was reduced to the dihydroquinone 389 by sodium dithionite. Treatment of 389 with boron tribromide and tetrabutylammonium iodide at 0 °C gave demethylated phenol 390, which was oxidized to 391 by aerobic oxidation. Chemoselective O-allylation of 391 with carbonate 392 catalyzed by Pd(PPh3)4 produced 393, which underwent regioselective Claisen rearrangement by heating at 80 °C on-water to generate murrayaquinone-E 82. Alkylation of 82 with methyl iodide and BuLi gave murrayaquinone-B 79. On the other hand, chemoselective O-allylation of 391 with carbonate 394 furnished 395, which was converted to murrayaquinone-D 81 by Claisen rearrangement. Similarly, alkylation of 81 with methyl iodide and BuLi delivered murrayaquinone-C 80 (Scheme 35).
Total Synthesis of Murrayaquinones -B-E 79-82 and Königinequinone A 385 by Norcott and McErlean.
Karan et al completed a short route to access glycozoline 75 and glycozolinol 396 in high yields.45 Treatment of N-protected indole 397 with crotyl boronic acid 398 followed by TBAF deprotection gave glycozoline 75. Removal of methyl ether in 75 by treatment with BBr3 afforded glycozolinol 396 (Scheme 36).
Total Synthesis of Glycozoline 75 and Glycozolinol 396 by Karan et al.
Polley et al accomplished the total synthesis of clausine-V 399 and clauszoline-K 220.46 The key step involves a palladium(II)-catalyzed C-H arylation of aniline carbamates with diazonium salts. Reaction between aniline carbamate 400 and diazonium salt 401 afforded 402, which underwent carbamate deprotection under basic conditions to give aniline 403. This aniline was protected as 404 with 2- picolinamide followed by the C-H amination reaction under inexpensive copper catalysis to furnish clausine V 399, For the synthesis of clauszoline K 220, carbazole 229 was synthesized from aniline carbamate 400 and diazonium salt 406 following the same route. DDQ oxidation of 229 then delivered clauszoline K 220 (Scheme 37). Clausine-V 399 and clauszoline-K 220 were obtained in 5 steps (54% overall yield) and 6 steps (59% overall yield), respectively.
Total Synthesis of Clausine-V 399 and Clauszoline-K 220 by Polley et al.
Momin et al demonstrated the synthesis of ekeberginine 196, murrayaquinone-A 350 and glycozoline 75.47 The synthesis of ekeberginine 196 started from carbazole 410. This carbazole was converted to triflate ester 411, which underwent Stille coupling with tributyl (3-methylbut-2-en-1-yl) stannane 412 in the presence of tetrakistriphenylphosphine palladium to yield prenylated carbazole 413. Methoxylation of compound 413 using excess of sodium methoxide then furnished ekeberginine 196. The synthesis of murrayaquinone A 350 employed carbazole 414 as the starting material. The aldehyde group in 414 was protected using propane-1,3-dithiol to produce compound 415 in 90% yield. PCC oxidation of 415 resulted in quinone 416, which was transformed into murrayaquinone-A 350 by treatment with Raney nickel. Glycozoline 75 was obtained from 3-methyl carbazole 417 in two steps. Bromination of 417 using NBS afforded bromide 418 in excellent yield. Methoxylation of this bromide by NaOMe in the presence of CuI then provided glycozoline 75 (Scheme 38).
Total Synthesis of Ekeberginine 196, Murrayaquinone-A 350 and Glycozoline 75 by Momin et al.
Nishiyama et al achieved the total synthesis of clausamine E 203 in eight steps from indole 419 with an 8.6% overall yield.48 Initially, NH-indole 419 was protected as 420 using SEMCl. Addition of vinyl magnesium bromide 421 to 420 yielded alcohol 422, which was then protected as methyl ether 423. Cross diene metathesis of 423 with methyl acrylate 424 generated enoate 425. Pd-catalyzed cyclocarbonylation of 425 in the presence of CO (1 atm) and tributyl(vinyl)tin produced carbazole 426, which was converted to triflate 427 using PhNTf2, NaH. Coupling reaction between this triflate with 2-methylbut-3-en-2-ol gave 428, which was transformed into clausamine E 203 by SEM removal promoted by TBAF (Scheme 39).
Total Synthesis of Clausamine E 203 by Nishiyama et al.
A series of naturally occurring carbazole alkaloid including mukonine 109, koenoline 428, murrayafoline A 202, murrayanine 429, mukoeic acid 430, glycoborine 116, clauszoline-K 220, glyozolicine 58, glycozoline 75, mukolidine 60, and mukoline 59 was synthesized by Bhatthula et al49
The synthesis of mukonine 109 started from 3-bromo-5-hydroxybenzoic acid 431. Methylation of both hydroxy and carboxyl group in 431 provided ester 432, which was converted to 434 by treatment with bis(pinacolato)diboron 433 under palladium catalyst. Suzuki-Miyaura cross-coupling reaction of 434 with o-bromonitrobenzene 435 gave 436, which underwent reductive cyclization to form mukonine 109. Reduction of mukonine by LiAlH4 afforded murrayafoline A 202, while DIBAL-H reduced mukonine 109 to koenoline 428. In addition, saponification of 109 with KOH in water and ethanol delivered mukoeic acid 430. Oxidation of koenoline 428 by MnO2 produced murrayanine 429. The same routes were applied to prepare glycoborine 116 and carbazole 268 from bromides 108 and 9, respectively. DDQ oxidation of 268 then furnished clauszoline K 259 (Scheme 40).
Total Synthesis of Mukonine 109, Koenoline 428, Murrayafoline A 202, Murrayanine 429, Mukoeic acid 430, Glycoborine 116, Clauszoline-K 220, Glyozolicine 58, Glycozoline 75, Mukolidine 60, and Mukoline 59 by Bhatthula et al.
Similarly, compound 520 was obtained from bromide 69 in two steps. Reductive cyclization of 250 resulted in glyozolicine 100 and glycozoline 117 in 76% total yield with ratio of 1:1. Oxidation of glyozolicine afforded mukolidine 102, which was reduced to mukoline 101 by NaBH4 (Scheme 40).
Wu et al reported the total synthesis of hydrocarbazolone alkaloid (1R,2R,3R)-3-hydroxy-1,2-dimethyl-1,2,3,9-tetrahydro-4H-carbazol-4-one 444.50 Initially, the Diels-Alder reaction of (silyloxyvinyl)indole 445 with dienophile 446 catalyzed by chiral holmium complex afforded 447 in excellent yield and excellent enantiomeric excess. Reductive removal of the acyl oxazolidone moiety in 447 by LiAlH4 delivered primary alcohol 448, which was converted to dimesylates 449 by treatment with MsCl. LiAlH4 reduction of 449 produced 450, which was transformed into 451 using copper chloride and cesium fluoride. Conversion of 451 to the TMS enol ether followed by oxidation with PhI(O2CCF3)2 provided 452. Finally, removal of the protecting group furnished the natural compound (1R,2R,3R)-3-hydroxy-1,2-dimethyl-1,2,3,9-tetrahydro-4H-carbazol-4-one 444 (Scheme 41).
Total Synthesis of (1R,2R,3R)-3-Hydroxy-1,2-Dimethyl-1,2,3,9-Tetrahydro-4H-Carbazol-4-one 444 by Wu et al.
Markad and Argade demonstrated a short route to synthesize indizoline 337.51 Alkylation of (E)-2-((1-(tert-butoxycarbonyl)-1H-indol-3-yl)methylene)succinate 154 by prenyl bromide 198 gave 453, which was converted to acid 454 by saponification. Triphosgene induced intramolecular acylation of 454 followed by methylation provided carbazole 456, which was reduced to primary alcohol 457 by DIBAL-H at −25 °C. Finally, oxidation of this alcohol by PCC furnished indizoline 337 (Scheme 42).
Total Synthesis of Indizoline 337 by Markad and Argade.
Nishiyama et al achieved the total synthesis of koeniginequinones A 458 and B 459 in 2016.52 The synthesis of koeniginequinone A started from indole 460. Reduction of ester 460 by LiAlH4 delivered primary alcohol 461, which was oxidized to aldehyde 462 by MnO2. Iodination of 462 using molecular iodine followed by treatment with BOMCl yielded 463, which underwent nucleophilic addition with prepenyl magnesium bromide 464 to generate alcohol 465. This alcohol was protected as TBS ether 466. One-pot cyclocarbonylation with CO catalyzed by [Pd] in the presence of tributyl(vinyl)tin, followed by deprotection with TBAF and oxidation furnished quinone 467. Finally, reductive removal of BOM group in 467 afforded koeniginequinone A 458. The same procedure was applied to prepare koeniginequinone B 555 from indole derivative 565 (Scheme 43).
Total Synthesis of Koeniginequinones A 458 and B 459 by Nishiyama et al.
Gaikwad et al completed the total synthesis of murrayanine 429 and mukonine 109 in a short route.53 Initially, benzoic acid 477 was converted to methyl ester 478. Buchwald-Hartwig coupling of bromide 478 with aniline afforded diaryl amine 114, which underwent palladium(II)- catalyzed oxidative cyclization to provide mukonine 109. Reduction of mukonine 109 using DIBAL then furnished murrayanine 429 (Scheme 44).
Total Synthesis of Murrayanine 429 and Mukonine 109 by Gaikwad et al.
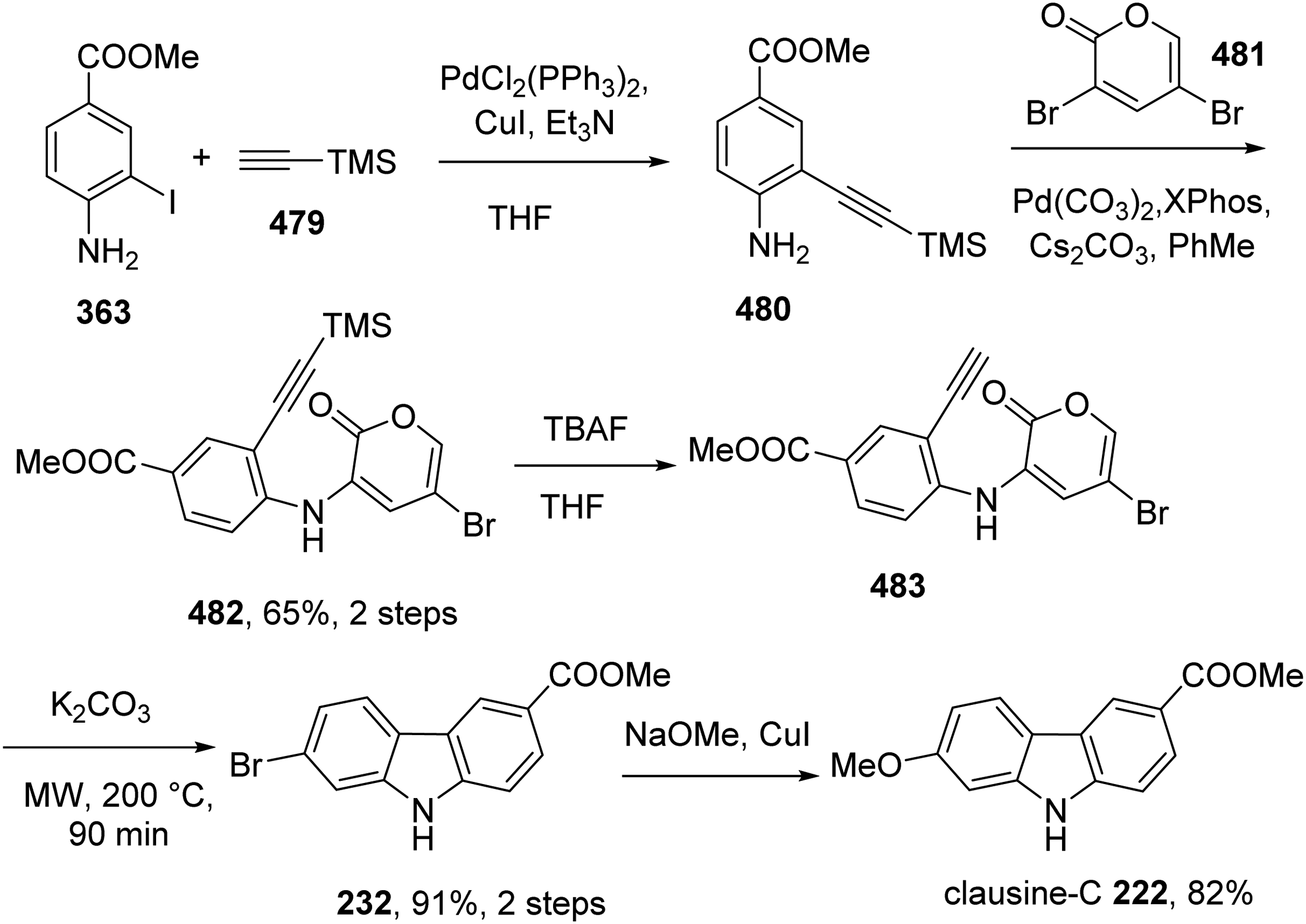
Points and Beaudry developed a new regioselective carbazole synthesis from 3-triflato-2-pyrones and alkynyl anilines and applied the method for the synthesis of clausine-C 222.54 Sonogashira cross-coupling reaction of iodide 363 with TMS-acetylene 479 produced 480. Amination of aniline 480 with 3,5-dibromo-2-pyrone 481 resulted in alkyne-tethered 3- anilido-2-pyrone 482, which was converted to 483 by cleavage of TMS group using TBAF. Intramolecular cycloaddition of 483 with the loss of CO2 furnished carbazole 232, which was transformed into clausine C by treatment with MeONa and CuI (Scheme 45). Clausine-C was obtained in 49% overall yield over five steps.
Total Synthesis of Clausine-C 222 by Points and Beaudry.
Recently, Okano group achieved the total synthesis of carbazomycins E 484 and F 485 based on double functionalization of an aryne intermediate generated from a 2-aminobiphenyl derivative.55 The synthesis started from 2-aminobiphenyl 486. Sequential treatment with 486 with BuLi and CuCN·2LiCl, DMF furnished carbazole 487. NH-carbazole 487 was protected as tosylate 488 before selective removal of one methyl ether affording 489. The hydroxy group in 489 was converted to triflate 490, which was then methylated with the formyl group intact using DABCO-AlMe3491 adduct, in the presence of palladium catalyst to generate compound 492. Removal of TS group from 492 gave 493, which underwent C4 demethylation using a combination of NaOH and dodecanethiol to furnish carbazomycin E 484. TBAF-mediated removal of Ts group in 597 gave 598, which was brominated by NBS to form 599. Methylation of 599 delivered carbazole 600, which was transformed to carbazomycin F 590 by treatment with dodecanethiol (Scheme 46).
The Okano group illustrated the total synthesis of carbazomycin A 152, carbazomycin B 153, carbazomycin C 496, carbazomycin D 49756 (Scheme 47) using their abovementioned procedure.55 Compound 486 was obtained from 498 by a sequence of iodination and Suzuki–Miyaura coupling. Sequential treatment with 603 with BuLi and CuCN·2LiCl, DMF provided carbazole 606. Selective removal of methyl at C2 using BCl3 delivered compound 607, which was then converted to triflate 608. Carbazomycin A was achieved in 95% yield on a gram scale by Negishi coupling of 608 with bis(trimethylaluminum)-1,4-diazabicyclo[2.2.2]octane 596. Treatment of carbazomycin A with dodecanethiol in the presence of NaOH at thermal conditionsn yielded carbazomycin B 199. Brromination of carbazomycin A using NBS in MeCN afforded bromide 609, which was converted to carbazomycin D 602 through Ullman reaction using NaOMe/CuI. Finaly, carbazomycin D 602 was converted to carbazomycin C 601 by treatment with dodecanethiol/NaOH (Scheme 47). The BCl3-promoted selective cleavage of methyl ether and Negishi coupling play the crucial role for the synthesis.
Total Synthesis of Carbazomycins E 484 and F 485 by Okano group.
Total Synthesis of Carbazomycins A-D by Okano group.
Recently, Pan et al demonstrated the total synthesis of carbazomycin A 152, carbazomycin D 497, and carbazomycin G 303 (Scheme 48).57 Initially, indole cyclopentanone 505 was transformed into carbazole 507 in gram scale by enolization, cyclopropanation, and expansion cascade. This is the vital step of the synthesis. Methylation of 507 with Me2SO4, K2CO3 afforded 508, which underwent SNAr reaction and Boc cleavage with NaOMe/CuI to furnish carbazomycin A (Scheme 48). An analogous fashion was preformed to prepare carbazomycin D 497 from indole cyclopentanone 509 (Scheme 48). Similarly, carbazole 513 was synthesized from indole cyclopentanone 512 by a sequence of enolization/ cyclopropanation/expansion. Oxidation dearomatization of 513 by PIDA produecd carbazoquinone 514, which was converted to 316 by methoxylation using MeONa/MeOH. Compound 316 could be transformed into carbazomycin G using literature report.35
Total Synthesis of Carbazomycin A 152, Carbazomycin D 497, and Carbazomycin G 303 by Pan et al.
Banerjee et al achieved the total synthesis of a series of natural carbazole alkaloids including carbazomycin A 152, carbazomycin D 497, hyellazole 154, 6- chlorohyellazole 155, sorazolon E 173, carazostatin 515, and lipocarbazoles A2-A4 516-518 (Scheme 49).58
Total Synthesis of Carbazomycin A 152, Carbazomycin D 497, Hyellazole 154, 6- Chlorohyellazole 155, Sorazolon E 173, Carazostatin 515, and Lipocarbazoles A2-A4 516-518.
Initially, the Brønsted acid-catalyzed cascade annulation between alkenyl indole 519 with glyoxal 520 gave sorazolon E 173, which was transformed into 521 by a sequence of methylation and Boc protection. Bromination of 521 using NBS, followed by Ullman reaction of the resulting bromide with NaOMe/CuI furnished carbazomycin A 152. On the other hand, methylation of 173, followed by double bromination resulted in compound 522. Treatment of 522 with NaOMe/CuI then produced carbazomycin D 497. Carazostatin 515 was obtained via the cascade annulation between alkenyl indole 523 with glyoxal. Carbazoles 172 and 524 were prepared from alkenyl indoles 252 and 253 using the same model, respectively. Methylation of these carbazoles then delivered hyellazole 154, 6-chlorohyellazole 155 (Scheme 49).
For the synthesis of lipocarbazoles A2-A4 516-518, reactions between 2-lithiated indole generated from indole 525 with aldehydes 526-528 provided indol-2-yl alcohols 529-531, respectively. Oxidation of these alcohols with IBX or DMP delivered ketones 532-534 in high yields. Removal of SO2Ph protecting group in 532-534 by TBAF provided 535-537 in excellent yields. Wittig reactions between ketone 535-537 with ylide 538 gave alkynyl indoles 539-541. Finally, the cascade annulation between alkenyl indoles 539-541with glyoxal under standard conditions furnished lipocarbazoles A2-A4 516-518 (Scheme 49).
The plausible reaction mechanism for the formation of 173 by the Brønsted acid-catalyzed cascade annulation between alkenyl indole 519 with glyoxal, the key step for this study, is illustrated at the end of Scheme 49. Initially, the nucleophilic addition of indole to glyoxal should first generate intermediate A, which underwent intramolecular nucleophilic addition through the alkene to form intermediate B. Subsequently, selective dehydration of B gives intermediate C, which is converted to carbazole 173 by aromatization.
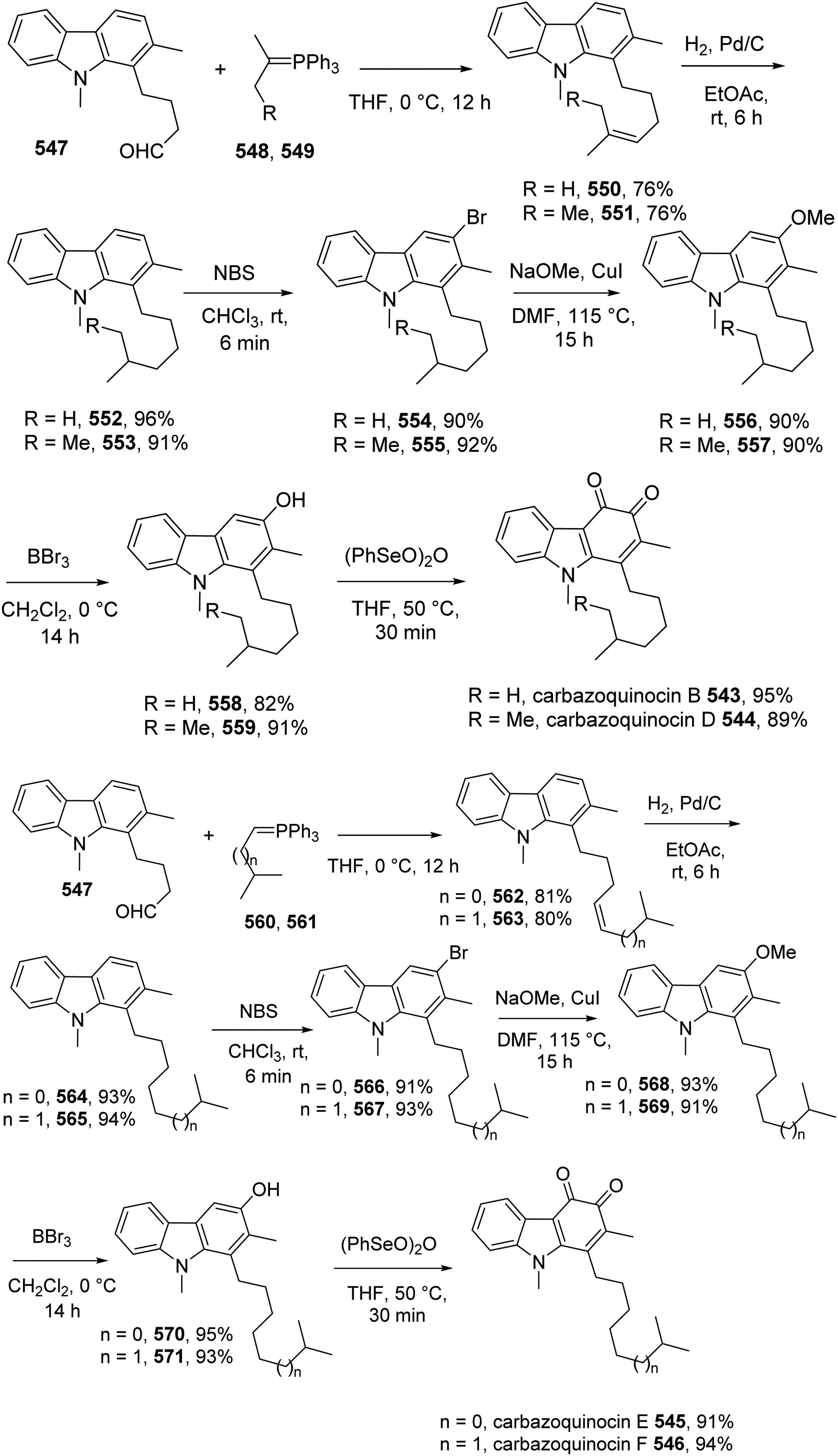
Bommanaboina et al introduced the multi-step synthesis of carbazoquinocins A 542, B 543, and D-F 544-546, carbazole alkaloids with long side chains at C1 (Scheme 63).59 The synthesis of carbazoquinocins B and D-F using methylindole 547 as the starting materials.
Wittig reactions between aldehyde 547 with phosphnium ylide 548 and 549 generated 550 and 551, respectively. Hydrogenation of 550 and 551 provided 552 and 553, which underwent bromination using NBS to yield bromides 554 and 555. Treatment of these bromides with NaOMe/CuI gave methoxylated products 556 and 557. Demethylation of 556 and 557 with BCl3 resulted in phenols 558 and 559, which underwent (PhSeO)2O-promoted oxidation to furnish carbazoquinocins B and D. The synthesis of carbazoquinocins E and F used the same procedure with the employment of phosphnium ylide 560 and 561 at the first step. Most steps of the synthesis were performed in excellent yields (Scheme 50).
Synthesis of Carbazoquinocins A 542, B 543, and D-F 544-546.
The synthesis of carbazoquinocin A started from ester 677. This ester was converted to alcohol 679 by a sequence of methylation and LiAlH4 reduction. This alcohol was oxidized to aldehyde 680, which underwent Wittig reaction with ylide 681 to afford 682. From here, the synthesis followed the same route for the construction of carbazoquinocins B and D-F (Scheme 63). Most steps of the synthesis were performed in excellent yields.
Nishiyama et al occomplised the total synthesis of mukoenine A 582 in seven steps.60 Firstly, the Wittig reaction between the indole-3-carbaldehyde 583 and ethyl 2-(triphenylphosphoranylidene)propionate 584 yielded α,β-unsaturated ester 585, which was reduced to primary alcohol 586 by DIBAL-H. Oxidation of 586 by MnO2 in DMF gave aldehyde 587, which took part in nucleophilic addition with fresly prepared organolithium compound 588 to generate alcohol 589. This alcohol was protected as MOM ether 590 before transforming into carbazole 592via allene-intermediate 591 by treatment with tBuOK. Finally, acid-catalyzed removal of two MOM protecting group in 592 furnished mukoenine A 582 (Scheme 51).
Total Synthesis of Mukoenine A 582.
Synthesis of Natural Fused Carbazole Alkaloids and Bis Carbazole Alkaloids
Synthesis of Natural Pyranocarbazole Alkaloids
In 2014, the Knölker group demonstrated the total synthesis of murrayacine 593, murrayacinine 594, 7-methoxymurrayacine 595, and clauraila E 596.8 The synthesis of intermediates 13, 21, 27, and 29 was presented in Scheme 1. Reduction of 13 by DIBAL-H in CH2Cl2 at −78 °C for 3.5 h gave murrayacine 593 in 80% yield. Under similar conditions, clauraila E 596, 7-methoxymurrayacine 594, and murrayacinine 701 were achieved from 21, 27, and 29 in 86%, 68%, and 87% yields, respectively. In these reduction reactions, ring opening products were also formed as minor isomers. When the reactions were performed at −78 °C and then −30 °C for specific times, the ring opening products were obtained as major isomers (Scheme 52).
Total Synthesis of Murrayacine 593, Murrayacinine 594, 7-Methoxymurrayacine 595, and Clauraila E 596 by Knölker group.
The group also demonstrated the total synthesis of seven natural pyranocarbazole alkaloids including pyrayafolines -A-E 597-601, O-methylmurrayamine A 602 and O-methylmahanine 603.9
Aniline 48 was obtained from nitro 604 by a sequence of TIPS protection and reduction. The Buchwald-Hartwig coupling of this aniline with bromide 22 afforded diarylamine 605, which was transformed into carbazole 606 by Pd-catalyzed oxidative cyclization. Removal of methyl ether in 606 by treatment with BBr3 provided 607. Reaction of 607 with carbonate 12 formed mixture of 608 (48% yield) and 609 (9% yield). TBAF-promoted removal of TIPS group in 608 and 609 provided pyrayafoline-C 599 and pyrayafoline-B 598, respectively. Methylation of pyrayafoline-C 599 then yielded pyrayafoline-A 597. Reaction of 607 with carbonate 28 formed mixture of 610 (49% yield) and 611 (9% yield). Similarly, cleavage of TIPS group in 610 and 611 using TBAF furnished pyrayafoline-D 600 and pyrayafoline-E 701, respectively (Scheme 53).
Total Synthesis of Pyrayafolines -A-E 597-601, O-Methylmurrayamine A 602 and O-Methylmahanine 603 by Knölker group.
TIPS deprotection of 606 resulted in 612, which was transformed into O-methylmurrayamine A 602 by reaction with prenal 613. In contrast, the reaction between 612 with citral 614 furnished O-methylmahanine 603 (Scheme 53).
The group established an efficient strategy for total synthesis of six pyranocarbazole alkaloids including (±)-mahanimbicine 615, murrayamine-J 616, bicyclomahanimbicine 617, murrayamine-M 618, murrayamine-G 619, and isomurrayazoline 620.61
Coupling reaction between p-toludine 67 with bromide 22 yielded diarylamine 621, which underwent oxidative cyclization to form carbazole 622. Removal of this carbazole by treatment with BBr3 follow by reaction with carbonate 28 and Claisen rearrangement provided (±)-mahanimbicine 615. Oxidation of 615 with DDQ produced murrayamine-J 616, while light-induced intramolecular [2 + 2] cycloaddition 615 delivered bicyclomahanimbicine 617. DDQ oxidation of 617 then afforded murrayamine-M 618. Finally, treatment of 615 with CSA for 11.5 day furnished a mixture of murrayamine-G 619 and isomurrayazoline 620 in 35% and 36% yields, respectively (Scheme 54).
Total Synthesis of (±)-Mahanimbicine 615, Murrayamine-J 616, Bicyclomahanimbicine 617, Murrayamine-M 618, Murrayamine-G 619, and Isomurrayazoline 620 by Knölker Group.
In another work, the group accomplished the total synthesis of murrayamine-A 624, mahanine 625, O-methylmahanine 603, murrayamines -B, -D, -H 626-628, and clauszolines -G, -B, and -H 629-631, mupamine 632, and heptazociline 633.62 The Buchwald-Hartwig coupling of the arylamine 36 and bromide 15 gave diaryl amine 634, which was converted to carbazole 635 by oxidative cyclization. Hydrolytic debenzylation of 635 provided 636, which reacted with prenal 613 in the presence of phenyl boronic acid to form pyranocarbazole 637. TBAF-catalyzed removal of TIPS group in 637 then delivered murrayamine-A 624. Meanwhile, reaction between 636 and citral 614 yielded pyranocarbazole 638, which was converted to mahanine 625 by TIPS removal using TBAF. Methylation of 625 using MeI, NaH then furnished O-methylmahanine 603. Treatment of 638 with TFA afforded 639, which underwent TIPS removal to give murrayamine-D 627 (Scheme 55).
Total Synthesis of Murrayamine-A 624, Mahanine 625, O-Methylmahanine 603, Murrayamines -B, -D, -H 626-628, and Clauszolines -G, -B, and -H 629-631, Mupamine 632, and Heptazociline 633 by Knölker Group.
Using the same procedure, pyranocarbazole 644 was achieved from arylamine 36 and bromide 640, while the use of arylamine 48 and bromide 132 resulted in pyranocarbazole 646. DDQ oxidation of 644 and 646 generated aldehydes 645 and 647, respectively, which were converted to clauszoline-G 629 and pyranocarbazole 648 by removal of the TIPS protecting group. Hydrogenation of 629 gave heptazociline 633. Methylation of 648 then provided clauszoline-B 630. Similarly, carbazole 651 was synthesized from arylamine 36 and bromide 68 in three steps. Reaction of this carbazole with prenal 613 delivered mupamine 632, while the use of citral 614 led to murrayamine-B 627. Finally, treatment of 627 with TFA afforded murrayamine-H 628 (Scheme 55).
In 2015, the group completed the total synthesis of murrayamine-A 624, O-methylmurrayamine-A 602, mahanine 625, O-methylmahanine 603, murrayamine-D 627, murrayamine-E 652, murrayamine-I 653, and murrayamine-K 654.63
The syntheses of murrayamine-A 624, mahanine 625, O-methylmahanine 603, and murrayamine-D 627 were following their previous report.62 Methylation of murrayamine-A 624 gave O-methylmurrayamine-A 602 in excellent yield. Treatment of 638 with CSA gave 655, which was converted to murrayamine-E 652 by TIPS removal using TBAF. Reaction between carbazole 184 with aldehyde 656 in the presence of Ti(O-iPr)4 provided pyranocarbazole 657, which underwent acetylation to furnish murrayamine-K 654. While reaction between carbazole 636 with aldehyde 656 in the presence of PhB(OH)2 delivered 7658, which then underwent TIPS cleavage to form murrayamine-I 653 (Scheme 56).
Total Synthesis of Murrayamine-A 624, O-Methylmurrayamine-A 602, Mahanine 625, O-Methylmahanine 603, Murrayamine-D 627, Murrayamine-E 652, Murrayamine-I 653, and Murrayamine-K 654 by Knölker Group.
The group also completed the total synthesis of pyranocarbazole alkaloids, glycomarrin 659 and eustifoline-B 660, along with simple carbazole alkaloids glycomaurrol 661 and eustifoline-C 662 by reduction of the pyranocarbazole alkaloids.12
The synthesis used glycozolinine 76, which was prepared in Scheme 4, as the key intermediate. Reaction between 76 and carbonate 12 afforded glycomarrin 659. Reduction of 659 using DIBAL-H in the presence of ZnCl2 then provided glycomaurrol 661. Meanwhile, reaction of between 76 and carbonate 28 delivered eustifoline-B 660, which was reduced by DIBAL-H to eustifoline-C 662 under similar conditions (Scheme 57).
Synthesis of Glycomarrin 659 and Eustifoline-B 660, Glycomaurrol 661, and Eustifoline-C 662 by Knölker Group.
In 2016, the group achieved the total synthesis of pyrayaquinones- A-C 663-665.13 The syntheses employed key intermediates 88, 89, and 91, which were prepared in the Scheme 5. LiAlH4-mediated deprotection of the tosyl group in 91 afforded the hydroxycarbazole 666, which was oxidized to pyrayaquinone-B 664 by using Frémy's salt. Bromination of 88 using NBS resulted in 667, which underwent Suzuki–Miyaura coupling with prenylboronic acid pinacol ester 668 to yield prenylated carbazole 669. Treatment of 669 with pyridinium chloride resulted in cleavage of the methyl ether and subsequent pyran ring closure and 670 was obtained in 80% yield. Ts deprotection of 670 byLiAlH4 followed by oxidation with Frémy's salt furnished 672, which was converted to pyrayaquinone-A 663 by DDQ oxidation. Reaction between hydroxy carbazole 89 and carbonate 28 gave ether 95, which underwent Claisen rearrangement to afford 786. Removal of the Ts protecting group in 673 then provided 674, which was oxidized to pyrayaquinone-C 665 by Frémy's salt (Scheme 58).
Total Synthesis of Pyrayaquinones- A-C 663-665 by Knölker Group.
In the same year, the group accomplished the total synthesis of koenine 675, koenimbine 676, 6,8-dimethoxygirinimbine 677, koenigine 678, and koenigicine 679.27
The synthesis of koenine 675 and koenimbine 676 was started from the intermediate 105, which was prepare in Scheme 6. BBr3-mediated methyl ether deprotection of 105 afforded 2-hydroxycarbazole 680, which reacted with prenal 613 to form pyranocarbazole 681. TBAF-promoted TBDPS deprotection of 681 provided koenine 675, which could be converted to koenimbine 676 by methylation. Alternatively, the reaction of carbalexin C 102 with prenal 613 delivered koenimbine 676 (Scheme 59). The synthesis of carbalexin C 102 was also presented in Scheme 6.
Buchwald-Hartwig coupling of bromide 682 with aniline 36 produced diarylamine 683, which was transformed into carbazole 684 by oxidative cyclization. Hydrolytic removal of the benzyl group in 684 followed by reaction with prenal 613 in the presence of PhB(OH)2 gave pyranocarbazole 686. TIPS deprotection of 686 using TBAF furnished koenigine 678, which was converted to koenigicine 679 by methylation. Using the same procedure, carbazole 689 was obtained form bromide 687 with aniline 36. Removal of the Bn group in 689 yielded 2-hyroxy carbazole 690, which was transformed into 6,8-dimethoxygirinimbine 677 by a sequence of ether formation with propargyl alcohol 19 and Claisen rearrangement. Alternatively, the reaction of 2-hyroxy carbazole 690 with prenal 613 furnished 6,8-dimethoxygirinimbine 677 (Scheme 59).
Total Synthesis of Koenine 675, Koenimbine 676, 6,8-Dimethoxygirinimbine 677, Koenigine 678, and Koenigicine 679 by Knölker group.
In 2018, the group described the first total synthesis of 7-isovaleryloxy-8-methoxygirinimbine 691 in 38.4% overall yield in 10 steps.64 The synthesis started from bromide 692. Methylation of 692 gave 693, which was converted to 694 by a sequence of Bayaer-Villiger oxidation and acid hydrolysis. Phenol 694 was protected as TIPS ether 695 before coupling with amine 36 to provide diarylamine 696. Pd-catalyzed oxidative cyclization of 696 yielded carbazole 697, which underwent benzyl deprotection by hydrogenation to form 698. Reaction between 698 and prenal 613 in the presence of PhB(OH)2 furnished pyranocarbazole 699. Removal of TIPS protecting group in 699 generated 700, which was transformed into 7-isovaleryloxy-8-methoxygirinimbine 691 by treatment with isovaleryl chloride 701 (Scheme 60).
Total Synthesis of 7-Isovaleryloxy-8-Methoxygirinimbine 691 by Knölker Group.
In 2020, the group completed the first total synthesis of clausenalansine A 702.65 The synthesis of pyranocarbazole 794 was followed Schemes 11, although different yields in steps were reported. Oxidation of 794 provided aldehyde 795, which was converted to clausenalansine A 814 by TBDPS removal using TBAF (Scheme 61). Clausenalansine A was avhieved in 29.7% overall yield over six steps.
Total Synthesis of Clausenalansine A 702 by Knölker Group.
Hou et al described an efficient approach for the total synthesis of 7-hydroxymurrayazolinine 704, murrayamine-D 627, and mahanine 625.66 The synthesis employed methyl carbonate 705 as the starting material. Base-catalyzed hydrolysis of 705 gave phenol 706, which reacted with propargyl alcohol 707 to form ether 708. Claisen rearrangement of 708 under microwave irradiation afforded chromene 709, which was converted to azide 710 by a sequence of reduction, diazotization, and azide displacement. Coupling reaction between 710 with boronic acid 711 produced azide diaryl 712. Cyclization of 712 under thermal conditions yielded pyranocarbazole 713, which underwent TBDMS removal to form mahanine 625. Finally, TsOH-catalyzed cyclization of 625 furnished a mixture of murrayamine-D 627 and 7-hydroxymurrayazolinine 704 in 45% and 37% yield, respectively (Scheme 62).
Total Synthesis of 7-Hydroxymurrayazolinine 704, Murrayamine-D 627, and Mahanine 625 by Hou et al.
Polley et al demonstrated the total synthesis of a series of pyranocarbazole alkaloids including girinimbine 714, murrayacine 593, mahanimbine 715, koenimbine 676, mahanine 625, murrayamine-D 627, and mukoenine-C 624.22 Mukoenine-C is another name for murrayamine-A. Reaction between aniline 716 and methyl chloroformate 717 generated 718, which was protected as benzyl ether 719. Bromination of 719 with NBS followed by basic hydrolysis gave 721. Suzuki-Miyaura cross-coupling reaction of 721 with boronic acid 722 delivered 723, which was converted to azide 724 by treatment with TMSN3 in the presence of t-BuONO. The mild, metal-free intramolecular C-N coupling reaction of 724 furnished carbazole 635, which underwent hydrolytic debenzylation to yield 636. Reaction of 636 with prenal 613 afforded pyranocarbazole 637, which was converted to mukoenine-C 624 (murrayamine-A) by removal of TIPS group. In addition, reaction of 636 with citral 614 provided pyranocarbazole 638. TBAF-induced removal TIPS group in 638 furnished mahanine 625, which was transformed into murrayamine-D 627 by treatment with TsOH (Scheme 63).
Total Synthesis of Girinimbine 714, Murrayacine 593, Mahanimbine 715, Koenimbine 676, Mahanine 625, Murrayamine-D 627, and Mukoenine-C 624 by Polley et al.
The synthesis of other compounds started from the intermediate 184, which was prepared in the Scheme 14. Reaction between 184 with prenal 613 delivered girinimbine 714, which was oxidized to murrayacine 593 by DDQ. Meanwhile, reaction of 184 with citral 614 gave mahanimbine 715. Similarly, reaction between carbalexin-C 102 prepared in the Scheme 6 with prenal 613 produced koenimbine 676 (Scheme 63).
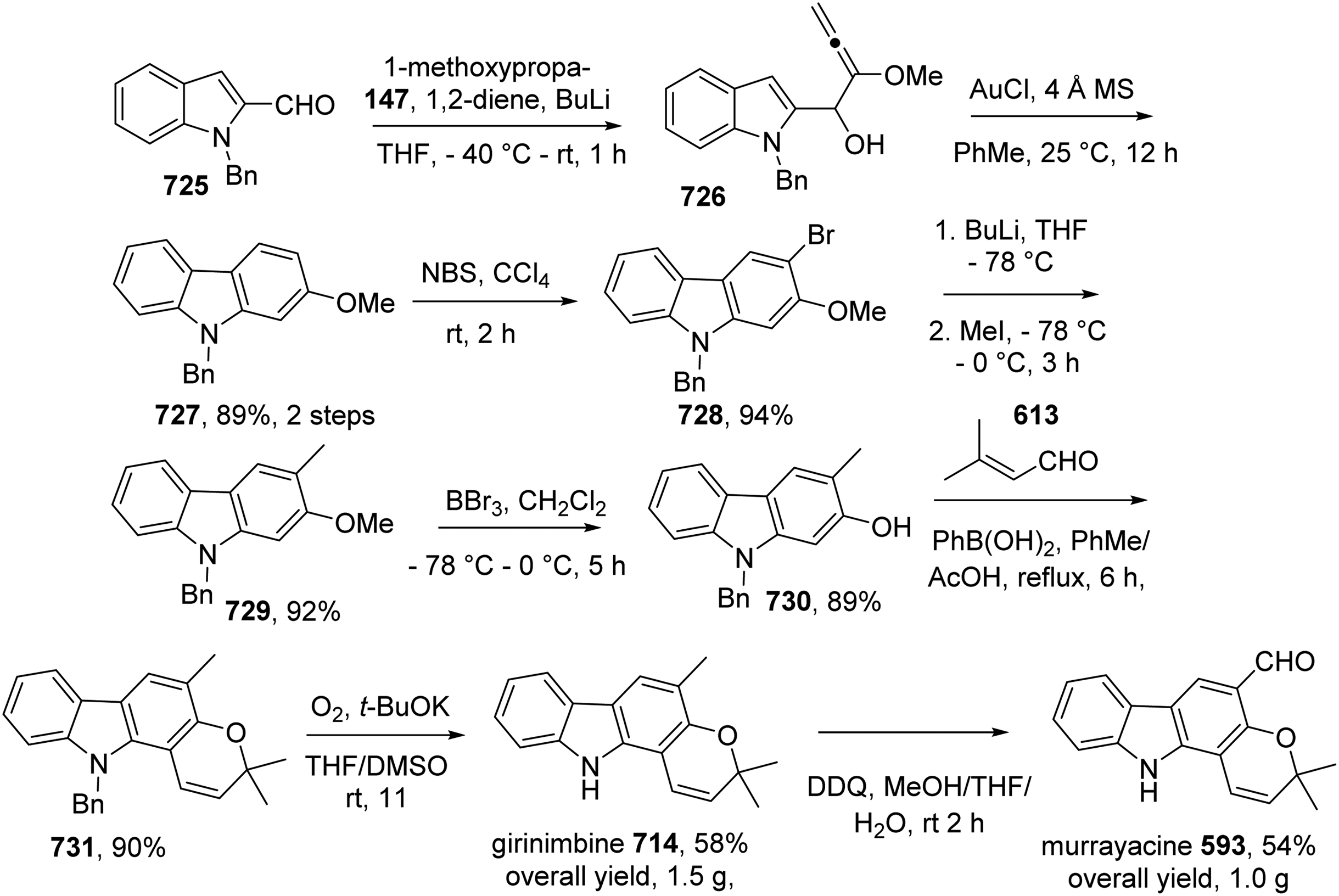
Dai et al presented the gram-scale total synthesis of girinimbine 714 and murrayacine 593 in 58% and 54% overall yields, respectively.67 The preparation of N-benzyl carbazole 728 from indole 725 and 147 in three steps was following previous report (Scheme 11),19 although yield of each step was improved. Treatment of 728 with MeI, BuLi gave 729, which was converted to 730 by BBr3-promoted demethylation. Reaction of 730 with prenal afforded pyranocarbazole 731, which underwent debenzylation to provide girinimbine 714 in 94% yield. Finally, oxidation of 714 by DDQ furnished murrayacine 593 (Scheme 64).
Total Synthesis of Girinimbine 714 and Murrayacine 593 by Dai et al.
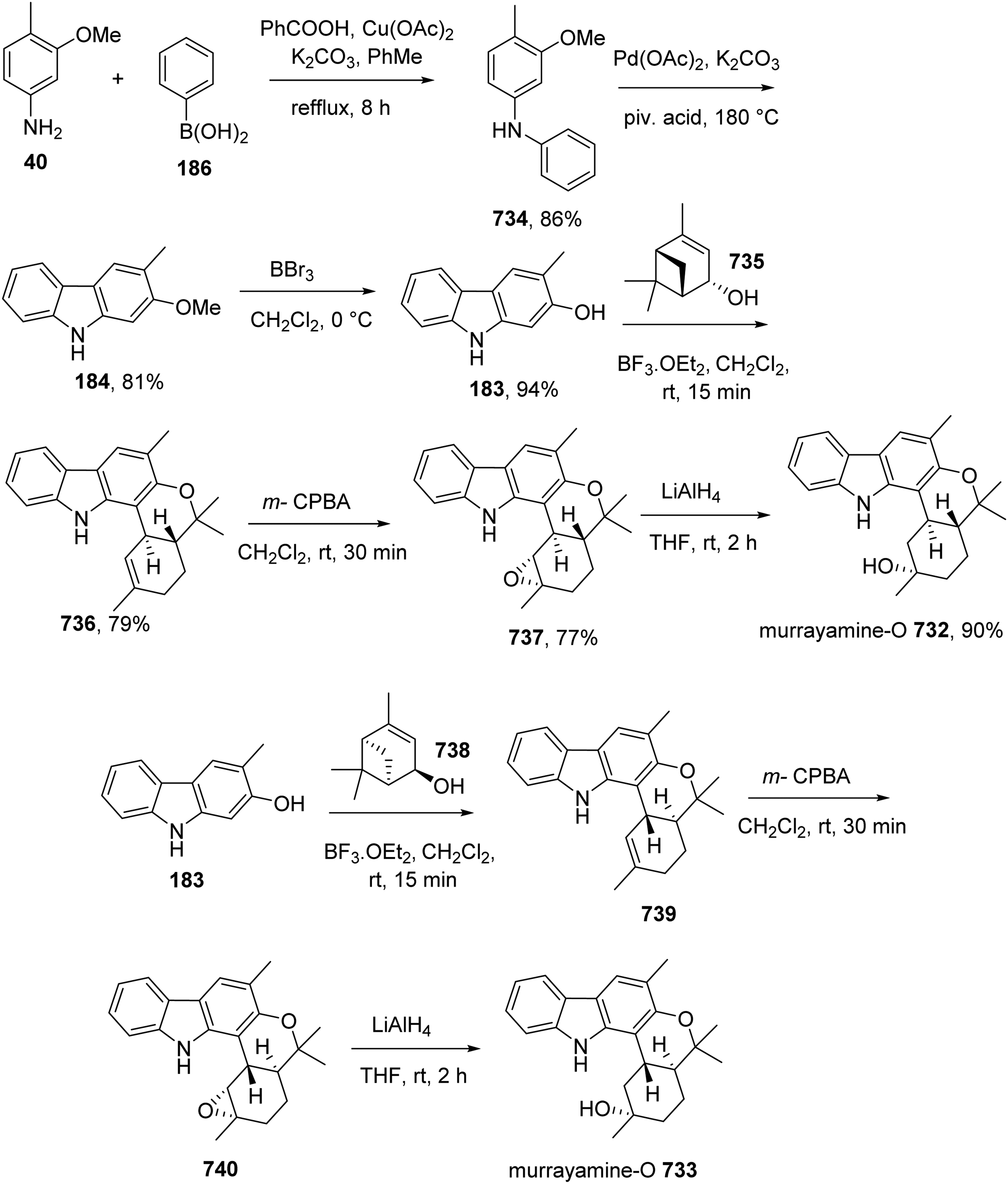
Dethe et al accomplished the first enantiospecific total syntheses of carbazole alkaloids murrayamines-O 732 and -P 733.68 Phenyl boronic acid 186 reacted with aniline 40 catalyzed by Cu(OAc)2 in refluxing toluene in the presence of K2CO3 afforded diarylamine 734, which was transformed into carbazole 184 by oxidative cyclization. Removal of the methyl ether in 184 generated 2-hydroxy-3-methyl carbazole 183. Reaction between 183 and (-)-(S)-cis-verbenol 735 in the presence of BF3·OEt2 furnished the pentacyclic compound 736. Epoxidation of 736 provided 737, which was reduced to murrayamine-O 732 by LiAlH4. A similar fashion was performed to prepare murrayamine-P 733 from 183 and (+)-(R)-cis-verbenol 738 (Scheme 65).
Total Synthesis of Murrayamines-O 732 and -P 733 by Dethe et al.
Reddy et al accomplished the total synthesis of 7-methoxyglycomaurin 741 and clauraila C 742.69 The synthesis started from resorcinol 743. Nitration of 743 afforded 4-nitroresorcinol 744, which was transformed into nitrochromene 746 by a sequence of etherification with propargyl chloride 745 and Claisen rearrangement. 746 was converted to triflate 747, which then underwent Suzuki-Miyaura cross coupling with boronic acid 384 to form 748. Cadogan reaction of 748 by treatment with PPh3 in DMAc at 150 °C furnished 7-methoxyglycomaurin 741. The synthesis of clauraila C 846 was followed the same route from triflate 851 and boronic acid 853 (Scheme 66).
Total Synthesis of 7-Methoxyglycomaurin 741 and Clauraila C 742 by Reddy et al.
Abbasa et al completed the first total synthesis of (±)-mafaicheenamine A 751 and the synthesis of clausine-E 752.70 The synthesis employed aniline 753 as the starting material. N-arylation of 753 with phenylead triacetate generated diarylamine 754, which underwent oxidative cyclization to provide carbazole 755. Hydrolytic removal of the benzyl group in 755 yielded clausine-E 752, which reacted with carbonate 756 forming 757. Claisen rearrangement of 757 under microwave irradiation produced prenylated carbazole 758. Acid 760 was the obtained from 758 by a sequence of methylation and based promoted ester hydrolysis. Finally, treatment of 760 with m-CPBA in CH2Cl2 followed by work-up with saturated NaHCO3 furnished (±)-mafaicheenamine A 751 (Scheme 67).
Total Synthesis of (±)-Mafaicheenamine A 751 and Clausine E 752 by Abbasa et al.
Norcott and McErlean achieved the total synthesis of pyrayaquinones -B 664 and -C 665 by oxidation.44 DDQ oxidation of prenylated carbazole alkaloids murrayquinones -E 82 and -D 81 in toluene without using a catalyst delivered pyrayaquinones -B 664 and -C 665, respectively (Scheme 68). Their synthesis of murrayquinones -D 81 and -E 82 was displayed in Scheme 35.
Synthesis of Pyrayaquinones -B 664 and -C 665 by Norcott and McErlean.
Polley et al reported the total synthesis of O-methylmahanine 603 based on palladium-catalyzed ortho C-H arylation of aniline carbamates with diazonium salt as the key step.46 Reaction between aniline carbamate 761 and diazonium salt 762 afforded 763, which underwent carbamate deprotection under basic conditions to give aniline 764. This aniline was converted to azide 765 by one-pot diazotization-azidation. Heating this azide at reflux toluene produced carbazole 766via nitrene insertion to the C-H bond. Hydrolytic debenzylation of 767 gave 2-hydroxycarbazole 754, which was transformed into O-methylmahanine 714 by reaction with citral 709 in the presence of Ti(OiPr)4 (Scheme 69).
Total synthesis of O-methylmahanine 603 by Polley et al
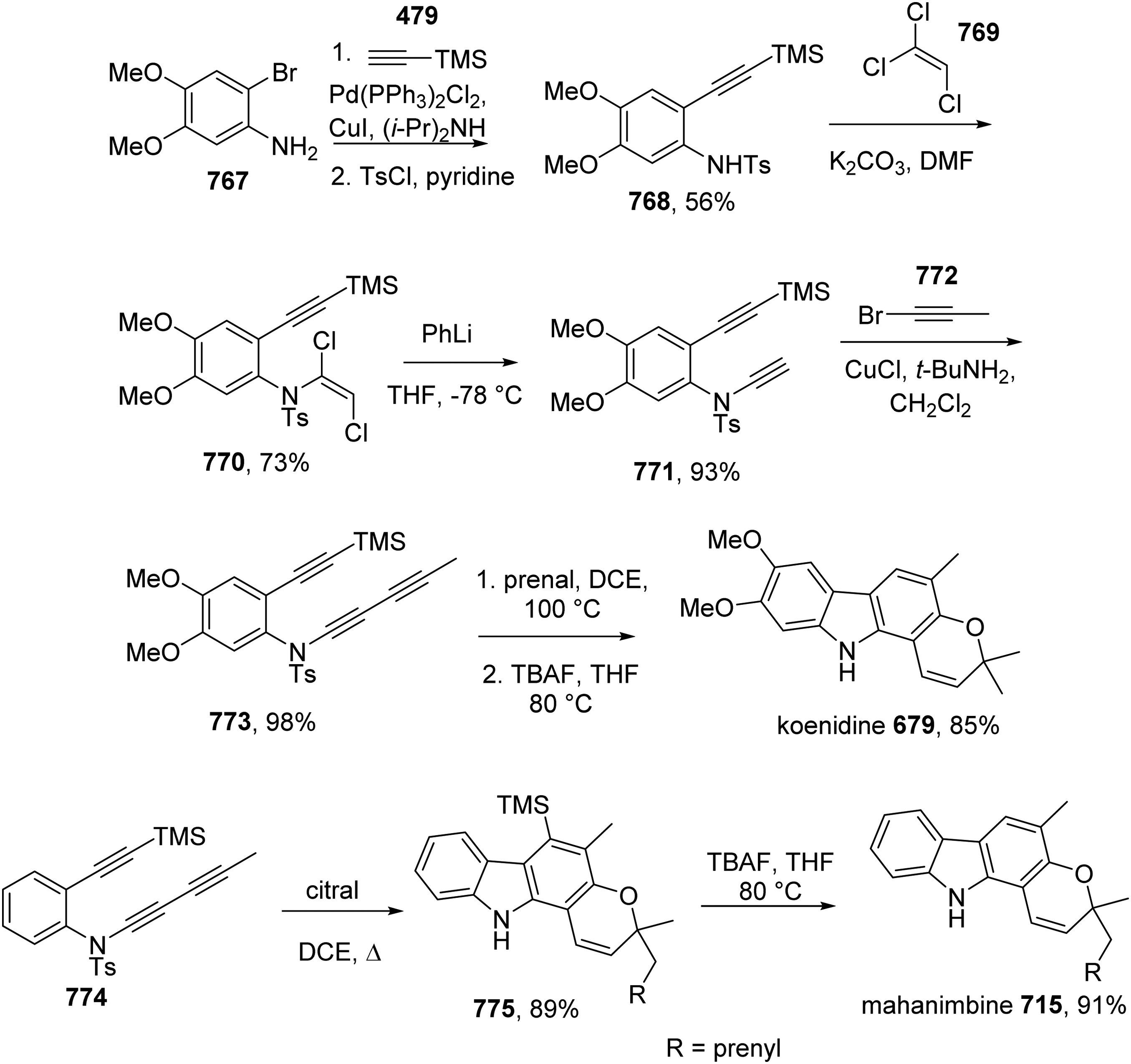
Wang and Hoye introduced an efficient route for the total synthesis of mahanimbine 715 and koenidine 679.71 Koenidine is also known as koenigicine. Compound 767 was converted to 768 by a sequence of Sonogashira coupling with alkyne 479 and tosylation. Reaction between tosylate 768 and trichloroethene 769 afforded 1,2-dichlorovinylsulfonamide 770. Dechlorination of 770 produced terminal alkyne 771, which underwent cross diyene coupling with alkynyl bromide 772 to yield triyne 773. The well-established hexadehydro-Diels-Alder cascade reaction of 773 by heating with prenal 613 then furnished koenidine 679. Meanwhile, the hexadehydro-Diels-Alder cascade reaction of 774 with citral 613 delivered 775, which was transformed in to mahanimbine 715 by TMS removal using TBAF (Scheme 70).
Total Synthesis of Mahanimbine 715 and Koenidine 679 by Wang and Hoye.
In 2017, Zang et al demonstrated the total synthesis of O-methylmurrayamine A 602 and 7-methoxymurrayacine 595.72 The synthesis started from nitro compound 776. Compound 106 was obtained from 776 by a sequence of TBDPS protection and nitro reduction. Buchwald-Hartwig coupling of bromide 22 with aniline 106 generated diarylamine 778, which was transformed into carbazole 779 by Pd-catalyzed oxidative cyclization. Removal TBDPS group in 779 by TBAF provided 2-hydroxycarbazole 612. The reaction of this carbazole with 3-methyl-2-butene 780 in the presence Ti(OiPr)4 then furnished O-Methylmurrayamine A 602. Finally, oxidation of 602 by DDQ delivered 7-methoxymurrayacine 595 (Scheme 71).
Total Synthesis of O-Methylmurrayamine A 602 and 7-Methoxymurrayacine 595 by Zang et al.
Markad and Argade accomplished the total synthesis of mafaicheenamine A 751, claulamine A 781, claulansine A 782, and the proposed claulamine E 783.51 The synthesis started from the intermediate 456, which was prepared in Scheme 42.51 Dihydroxylation of 456 with OsO4, NMO followed by lactonization provided mafaicheenamine A 751. Treatment of mafaicheenamine A with the Burgess reagent resulted in claulamine A 781 in 74% yield through dehydration process. Reduction of 751 by DIBAL-H produced claulansine A 782 in 77% yield. Oxidation of 456 with m-CPBA delivered proposed claulamine E 783 (Scheme 72). The structural assignment of 783 was confirmed by x-ray crystallography. However, the obtained analytical and spectral data for compound 783 was not in agreement with reported data for the natural product claulamine E.
Total Synthesis of Mafaicheenamine A 751, Claulamine A 781, Claulansine A 782, and the Proposed Claulamine E 783 by Markad and Argade.
Nishiyama et al performed the synthesis of girinimbine 714 from mukoenine A by the construction of the pyran ring via selenoetherification of the o-allylic phenol moiety (Scheme 73).60 The synthesis of mukoenine A is outline in Scheme 51.
Total Synthesis of Girinimbine 714 by Nishiyama et al.
Lu et al achieved the total synthesis of six natural pyrano[3,2-a]carbazole alkaloids, namely girinimbine 714, mukoenine-C 624, koenimbine 676, koenigine 678, koenine 675, and koenidine 679.73 Initially, alkylation of phenol 776 with 3-chloro-3-methyl-1-butyne 745 resulted in aryl propargyl ether 784, which underwent a sequence of thermally induced [3,3]-sigmatropic rearrangement, in situ rearomatization, and cyclization to give 5-nitrochromene 785. Reduction of 785 iron in glacial acetic acid afforded 5-aminochromene 786. Buchwald-Hartwig amination of 786 with bromobenzene delivered diaryl amine 787, which was converted to girinimbine 714 by an oxidative cyclization process. The syntheses of koenimbine 676, koenidine 679, mukoenine-C 624, koenigine 678, koenine 675 from 5-aminochromene 785 employed the same procedure with the use of bromides 77, 790, 792, 795, and 798, respectively (Scheme 74).
Total Synthesis of Girinimbine 714, Mukoenine-C 624, Koenimbine 676, Koenigine 678, Koenine 675, and Koenidine 679 by Lu et al.
Dangar et al described the total synthesis of mahanimbine 715, bicyclomahanimbine 801, murrayazolinine 802, curryanin 803 (also known as cyclomahanimbine), and isocyclomahanimbine 804.74 Firstly, bromide 805 was converted to boronic acid 384, which then underwent a Pd(0)-catalyzed Suzuki–Miyaura coupling with readily available bromide 806 to provide compound 807. Nitro 807 was reduced to aniline 808 before protecting as acetamide 809. Dehydrogenative electrochemical reaction of 809 produced carbazole 810, which then was converted to 183 by base-catalyzed hydrolysis followed by BBr3-promoted ether cleavage. Reaction of 183 with citral in pyrridine at elevated temperature furnished mahanimbine 715 in good yield. Photocatalytic cyclization of 715 using Cu(OTf)2 as a catalyst under the irradiation of a 500 W Na-lamp delivered bicyclomahanimbine 801 in high yield. On the other hand, curryanin 803 was achieved from 715 by photocatalytic cyclization using CSA as a catalyst under the irradiation of a 500 W Na-lamp. Murrayazolinine 802 was obtained from curryanin 803 through a sequence of epoxidation and LiAlH4 reduction. Finally, acid-catalyzed dehydration of 802 gave isocyclomahanimbine 804 (Scheme 75).
Total Synthesis of Mahanimbine 715, Bicyclomahanimbine 801, Murrayazolinine 802, Curryanin 803, and Isocyclomahanimbine 804 by Dangar et al.
Liu et al presented the synthesis of claulansine M 812.38 The synthesis started from carbazole 347, which was prepared in Scheme 29. [3,3]-sigmatropic rearrangement of propargyl ether 347 followed by rearomatization and cyclization provided 812 in excellent yield. Reduction of the ester group in 812 by LiAlH4 followed by DMP oxidation furnished claulansine M 812 (Scheme 76).
Total Synthesis of Claulansine M 812.
Synthesis of Other Fused Carbazole Alkaloids
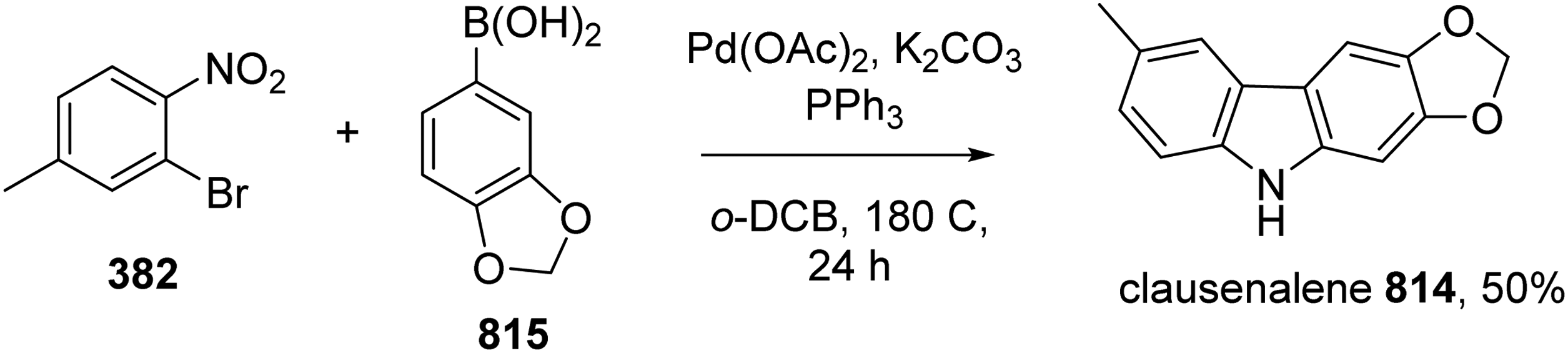
Rasheed et al developed an efficient method to synthesize clausenalene 814.41N-arylation of iodobenzoate 363 by boronic acid 815 catalyzed by Cu(OAc)2 in toluene under air provided diarylamine 816. Pd-catalyzed intramolecular C–H arylation of 816 produced carbazole 817, which was transformed into clausenalene 814 by treatment with LiAlH4 (Scheme 77).
Total Synthesis of Clausenalene 912 by Rasheed et al.
Goo and Woo completed the synthesis of clausenalene 814via a tandem C-C cross-coupling and reductive amination.43 Reaction between o-bromonitrobenzene 382 and boronic acid 815 directly furnished clausenalene 814 in one pot (Scheme 78).
Total Synthesis of Clausenalene 814 by Goo and Woo.
Markad and Argade accomplished the total synthesis of clausenaline D 818 with high efficiency.21 Allylation of 158 by successive treatment with LiHMDS and allyl bromide afforded 819, which was converted to acid 820 by saponification. The Ac2O-AcONa stimulated dehydrative intramolecular cyclization of 820 yielded acylated carbazole 821. OsO4 oxidation of the allyl side chain in 821 gave aldehyde 822. A one-pot p-TSA- mediated double acetyl removal of 822 followed by dehydrative intramolecular cyclization furnished furocarbazole 823, which was reduced to primary alcohol 824 by DIBAL-H. Finally, PCC oxidation of this alcohol delivered clausenaline D 818 (Scheme 79).
Total Synthesis of Clausenaline D 818 by Markad and Argade.
Liu et al demonstrated the total synthesis of clausenaline D 818 from carbazole 344, which was prepared in Scheme 29.38 Dihydroxylation of 344 with OsO4 followed by sodium periodate oxidation delivered aldehyde 825. Treatment of this aldehyde with BBr3 produced furocarbazole 826 via demethylation and concomitant furan ring formation. Clausenaline D 818 was achieved from 826 by a sequence of LiAlH4 reduction and DMP oxidation (Scheme 80).
Total Synthesis of Clausenaline D 818 by Liu et al.
Roy and Mal achieved the total synthesis of furanoclausamine B 827 and harmandianamine A 828 in a regiodefined manner from indole 829.24N-Boc protected of 829 generated 830, which was transformed into furoindolone 832 by the reaction with Grignard reagent 831 followed by cyclization. The annulation of this furoindolone with dimethyl fumarate 833 in the presence of LDA afforded carbazole 834. Compound 835 was achieved from 834 by a sequence of saponification, decarboxylation, and Boc protection. Bromination of 835 with NBS provided bromide 836, which was oxidized to aldehyde 837 by NMO in MeCN. Nucleophilic addition of Grignard reagent 838 to this aldehyde gave 839, which was converted to harmandianamine A 828 by heating at 120 °C to remove Boc groups. Finally, methylation of 828 with MeI/K2CO3 then produced furanoclausamine B 827 (Scheme 81).
Total Synthesis of Furanoclausamine B 827 and Harmandianamine A 828 by Roy and Mal.
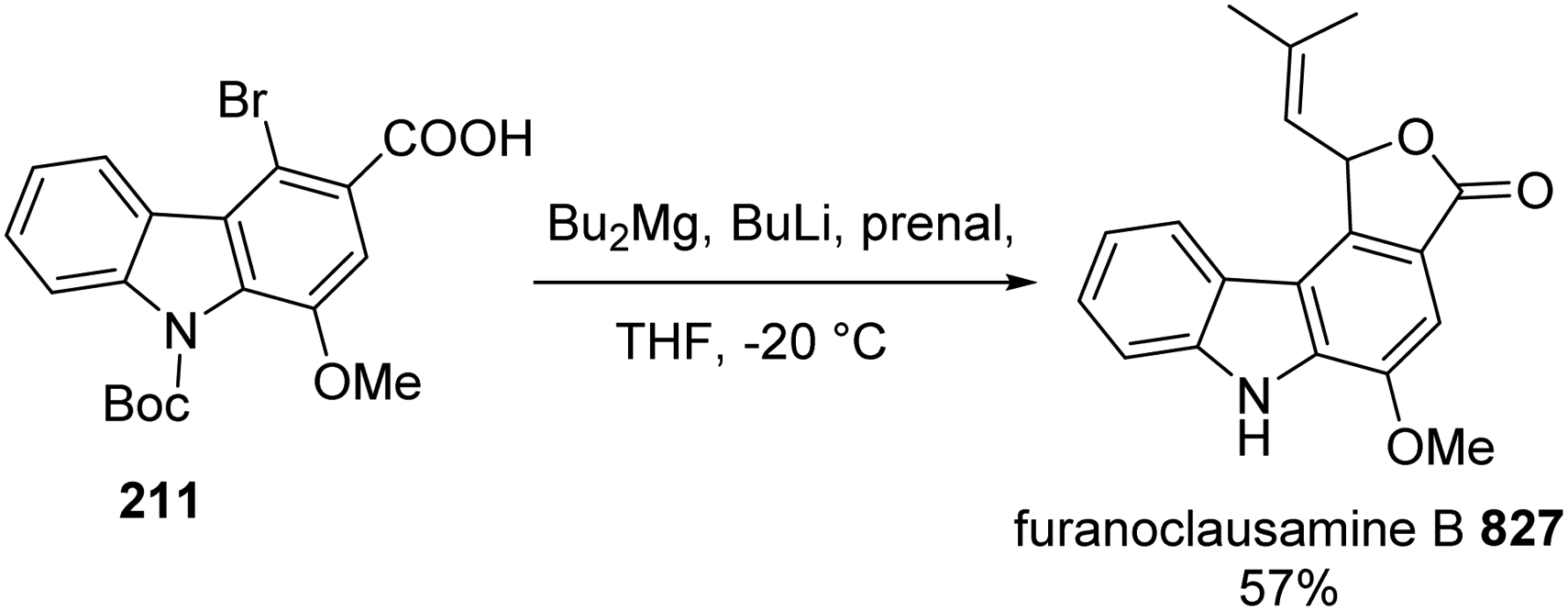
In 2015, these authors employed this approach with modification to synthesize furanoclausamine B 827.25 Treatment of bromide 211, which was prepared in the Scheme 16, with Bu2Mg, BuLi then prenal 613 furnished furanoclausamine B 827 (Scheme 82).
Total Synthesis of Furanoclausamine B 827 by Roy and Mal.
Spindler et al demonstrated the first enantioselective total synthesis and the assignment of the absolute configuration of furoclausine-B 840.75 Nitrophenol 604 reacted smoothly with 2-methylbut- 3-yn-2-yl trifluoroacetate, prepared in situ from the propargylic alcohol 19, to provide propargyl ether 841. Hydrogenation of the triple bond in 841 using Lindlar's catalyst afforded 842, which underwent Claisen rearrangement to form prenylated nitrophenol 843. TIPS protection of 843 yielded 844, which was transformed into dihydrobenzofuran 845 in excellent yield and significant enatiomeric excess by a sequence of Shi epoxidation and TBAF-mediated TIPS removal. Reduction of 845 by H2/PtO2 generated amine 846, which was converted to diarylamine 847 by Buchwald–Hartwig coupling with bromide 15. Palladium(II)-catalyzed oxidative cyclization of 847 gave carbazole 848, which was oxidized to aldehyde 849. Finally, removal of the TIPS group in 849 using TBAF furnished furoclausine-B 840 (Scheme 83).
Total Synthesis of Furoclausine-B 840 by Spindler et al.
Martín-Torres et al achieved the total synthesis of mafaicheenamine C 850 based on enantioselective alkoxycyclization of 1,6-enynes with Gold(I)- Cavitands as the key step.76 The synthesis started from carbazole 851. Reaction of 851 with the Bestmann–Ohira reagent 852 afforded 1,6-enyne 853. The chiral gold(I)-cavitand catalyzed -the enantioselective alkoxycyclization of 1,6-enyne 853 furnished 854 in excellent yield and high ee. Pd-catalyzed deallylation of 854 provided 855, which was transformed into mafaicheenamine C 850 by oxidative cleavage of the exocyclic alkene using OsO4, NaIO4 (Scheme 84).
Total Synthesis of Mafaicheenamine C 850 by Martín-Torres et al.
Mal and Roy completed the total synthesis of claulansine D 856 and mafaicheenamine E 857.77 Annulation reaction of furoindolone 858 with dimethyl maleate and LDA gave carbazole 859. Selective reduction of 859 with LiAlH4 at −78 °C afforded furocarbazole 860, which was converted to 861 by methylation. Bromination of 861 yielded 862, which was transformed into 863 by refluxing in dioxane/H2O. Alkylation of 863 with methallyl bromide 864 in the presence of indium produced 865, which isomerized to mafaicheenamine E 857 by heating with PTSA in toluene. N-Boc protected of 857 delivered 866, which was oxidized to diol 867. Finally, neat heating of 867 at 140 °C furnished claulansine D 856 (Scheme 85).
Total Synthesis of Claulansine D 856 and Mafaicheenamine E 857 by Mal and Roy.
Goetz et al described the enantiospecific total synthesis of (+)-tubingensin A 868.78 The synthesis started from chiral conjugate enone 869. Vinyl cuprate addition to enone 869 followed by trapping of the resulting enolate gave silyl enol ether 870 as a single diastereomer. Treatment of 870 with 9-BBN followed by Pd-catalyzed cross-coupling with carbazole 871 afforded 872. Compound 874 was obtained from 872 by treatment with salt 873 followed by m-CPBA oxidation. Cuprate addition of 875 to 874 and trapping the resulting enol with TESCl delivered 876. Treatment of 876 with NaNH2 generated 877, which converted to 878 by MOM cleavage. Finally, reduction of 878 by Na in i-PrOH furnished (+)-tubingensin A 868 (Scheme 86).
Total Synthesis of (+)-Tubingensin A 868 by Goetz et al.
Guo et al accomplished the total synthesis of lansine F 879 and clauemarazole D 880.79 The synthesis started from carbazole 881. Dihydroxylation of 881 followed oxidative cleavage of the resulting vicinal diol by sodium periodate afforded aldehyde 882. Treatment of this aldehyde with ammonium acetate provided pyrido[4,3-b]carbazol-1-one 883. Treatment of 883 with triflic anhydride followed by palladium-catalyzed reduction unstable triflate furnished lansine F 879 (Scheme 87).
Total Synthesis of Lansine F 879 and Clauemarazole D 880 by Guo et al.
On the other hand, hydroboration-oxidation of 881 gave primary alcohol 884, which was converted to ester 886 by a sequence of oxidation and methylation. Diester 886 was protected as N-Boc 887, which underwent Dieckmann condensation to generate tetracycle 888. Decarboxylation of 888 was achieved by treatment with DBU at elevated temperature to deliver 889. Finally, removal of Boc group in 889 produced clauemarazole D 880 (Scheme 87).
Ji et al accomplished the total synthesis of olivacine 890 from carbazole 341 in high yield.39 Allylation of 341 with allyl bromide gave 891, which was converted to triflate 892. Palladium-catalyzed cross coupling of this triflate with trimethylaluminum 893 produced 894. Dihydroxylation of terminal alkene 894 followed by oxidative cleavage afforded aldehyde 895. Reaction of this aldehyde with ammonium acetate in the presence of acetic acid delivered 896. Treatment of 896 with triflic anhydride followed by coupling of the resulting an unstable triflate with trimethylaluminum furnished olivacine 890 (Scheme 88).
Total Synthesis of Olivacine 890 by Ji et al.
In 2015, Ute Kober and Knölker completed the total synthesis of malasseziazole C 897.80 Condensation of dicarboxylate 898 with an excess of aniline 899 gave diethyl 2,5-bis(phenylamino)terephthalate 900, which underwent palladium(II)-catalyzed oxidative cyclization to generate diethyl 5,11-dihydroindolo[3,2-b]carbazole-6,12-dicarboxylate 901. Selective reduction of this carbodixylate produced the monoalcohol 902, which was oxidized to aldehyde 903. Finally, saponification of 903 furnished malasseziazole C 897 (Scheme 89). Malasseziazole C was afforded in 39% overall yield over five steps.
Total Synthesis of Malasseziazole C 897 by Ute Kober and Knölker.
In 2019, Dong et al demonstrated a formal [1 + 2 + 3] annulation of methyleneindolinones with o-alkenyl arylisocyanides for the synthesis of indolo[3,2-b]carbazoles. The approach was applied for the synthesis of malasseziazole C 897 in a short and efficient route.81 The annulation reaction between methyleneindolinone 904 and o-alkenyl arylisocyanide 905 furnished 5,11-dihydroindolo[3,2-b]carbazole-6,12-dicarboxylate 901. Reduction of this dicarboxylate provided a mixture of primary alcohol 902 and aldehyde 903. Oxidation of 902 gave 903 in excellent yield. The other ester group in 903 was finally hydrolyzed under basic conditions to provide malasseziazole C 897 (Scheme 90).
Total Synthesis of Malasseziazole C 897 by Dong et al.
Kaliyaperumal et al achieved the total synthesis of calothrixin B 904 starting from compound 376, which was prepared in Scheme 33.42 Pd(OAc)2-catalyzed intramolecular C–X/C–H cross coupling reaction of 376 provided carbazole 380. The reductive amination of 380 with 2-iodoaniline 905 was performed using sodium triacetoxyborohydride in the presence of TFA to generate 906. N-acylation of 906 with acetyl chloride gave 907, which was transformed into 908 by Pd(OAc)2-catalyzed intramolecular C–X/C–H cross coupling. Oxidation of 908 by CAN then furnished calothrixin B 904. Alternatively, reductive amination of 376 with 2-iodoaniline 905 resulted in 909, which underwent N-acylation to form 910. The double intramolecular C–X/C–H cross coupling reaction of 919 using Pd(OAc)2 as catalyst in the presence of PCy3 and of JohnPhos furnished 908, which was oxidized to calothrixin B 994 by CAN (Scheme 91). Calothrixin B was achieved in 64% overall yield over five steps from 376.
Total Synthesis of Calothrixin B 904 by Kaliyaperumal et al.
Saravanan et al demonstrated the total synthesis of calothrixins A 911 and B 904 from 2-methyl indole 912.82 Selective N-phenylsulfonylation of this indole using N-phenylsulfonylbenzotriazole 913 and NaH gave 914. Acylation of 914 with propionic anhydride 915 using BF3·OEt2 as the catalyst at 0 °C afforded 916, which was converted to bromide 917 by bromination with NBS at the methyl side chain. The reaction of bromide 917 with triphenylphosphine, followed by Wittig reaction of the resulting phosphonium salt with 2-nitroaryl aldehyde 918 produced 2-nitroarylvinyl-3-propionyl indole 919, which was converted to 920 by α-bromination. Reaction of 920 with phenyl sulfinate 921 yielded the desired sulfone 922, which underwent a sequence of O-methylation with dimethyl sulfate and thermal electrocyclization of the resulting trienes in xylenes at reflux to afford carbazole 923. Benzylic bromination of 923 generated 924, which was oxidized with bis(tetrabutylammonium) dichromate (TBAD) in refluxing CHCl3 to form 925. Compound 927 was obtained from 925 by a sequence of BBr3-mediated methyl ether deprotection and desulfonylation with NaOH. Finally, reductive cyclization of 927 in the presence of Pd/C under H2 atmosphere, followed by oxidation of the resulting 13-hydroxyquinocarbazoles using CAN in furnished calothrixins A 911 and B 904 in a 1:1 ratio (Scheme 92). In most steps, products were obtained in good to excellent yields.
Total Synthesis of Calothrixins A 911 and B 904 by Saravanan et al.
Ramkumar and Nagarajan designed an efficient route for the total of olivacine 890 and calothrixin B 904.83 The synthesis employed ethyl pyrrole-carboxylate 932 as the starting material. Treatment of 932 with 2-methylnicotinaldehyde 933 in the presence of 1,1,3,3,-tetramethylguanidine (TMG) followed by IBX oxidation gave ketone 934. Wolff-Kishner reduction of this ketone produced compound 935, which was transformed into 936 by treatment with LDA and HMPA. Addition of MeMgI into 936 followed by treatment with NaBH4/AlCl3 in dry THF afforded olivacine 890. Similarly, reaction between 932 and quinoline-3-carboxaldehyde 937 catalyzed by TMG in MeOH provided alcohol 938, which was converted to 939 by DMP oxidation. Finally, treatment of 939 with LDA and HMPA furnished calothrixin B 904 (Scheme 93).
Total Synthesis of Olivacine 890 and Calothrixin B 904 by Ramkumar and Nagarajan.
These two authors introduced another approach towards the total synthesis of calothrixin B 904 from compound 940.84 Reduction of 940 by LiAlH4 delivered 941, which was oxidized to 942 using CAN. Suzuki cross-coupling reaction of 942 with boronic acid 943 generated 944, which was transformed into compound 946 by copper-catalyzed domino reaction with benzyl amine 945. Finally, N-deprotection of the derivative 946 using AlCl3 in anisole at 100 oC furnished calothrixin B 904 (Scheme 94).
Total Synthesis of Calothrixin B 904 by Ramkumar and Nagarajan.
In another report, these authors synthesized calothrixin B 904 in five steps from commercially available starting materials.85 The Sonogashira coupling reaction of 2-ethynyl-1H-indole 947 with iodide 948 resulted in 949. Copper-catalyzed oxidative cyclization reaction of enyne 949 afforded 950, which underwent base catalyzed hycrolysis to provide acid 951. Acid catalyzed cyclization of 951 gave 952, which was converted to calothrixin B 904 by treatment with 10% Pd/C and HCOONH4 in MeOH at reflux (Scheme 95). Calothrixin B was synthesized in 41.5% overall yields over five steps.
Total Synthesis of Calothrixin B 904 by Ramkumar and Nagarajan.
Mori-Quiroz et al designed a short and efficient route for the synthesis of calothrixins A 911 and B 904 from murrayaquinone A 350.86N-Boc protection of 350 yielded 953, which underwent bromination with NBS to give bromide 954. Reaction between 954 with aniline 955 provided secondary amine 956, which was oxidized to 957 by Ag2O. Intramolecular Suzuki coupling reaction of 957 furnished calothrixin B 904, which was oxidized to calothrixin A 911 by m-CPBA. Calothrixin B was achieved in 5 steps and 45% overall yield (Scheme 96).
Total Synthesis of Calothrixins A 911 and B 904 by Mori-Quiroz et al.
Lee et al achieved the total synthesis of arcyriaflavin A 958 and calothrixin B 904 starting from indole 959.87 Treatment of 959 with NaCN in DMF at 60 °C produced 2,2′-bisindole-3-acetic acid 960 in excellent yield. N-benzylation of 960 resulted in 961, which underwent Friedel-Crafts with ethyl glyoxylate 962 to generate 963. Oxidation of 963 afforded 3′-α-ketoester 964, which was converted to indolocarbazole 965 by treatment with t-BuOK. Compound 965 was subjected to hydrolysis under basic conditions to delivered dicarboxylic acid, which then underwent dehydration to furnish anhydride 966. Reaction of this anhydride with NH4OAc at elevated temperature produced imide 967, which was transformed into arcyriaflavin A 958 by AlCl3-mediated debenzylation. For the synthesis of calothrixin B 904, compound 961 was hydrolyzed to acid 968. Treatment of this acid with POCl3 in CHCl3 at 0 °C furnished 5-hydroxyindolecarbazole 969, which was transformed into 970 by Vilsmeier-Haack reaction. MOM protection of 970 provided 971, which was oxidized to 972 using CAN. Finally, cleavage of the benzyl group in 972 using AlCl3 in the presence of anisole delivered calothrixin B 904 (Scheme 97). Arcyriaflavin A was synthesized in 58% overall yields over eight steps.
Total Synthesis of Arcyriaflavin A 958 and calothrixin B 904 by Lee et al.
Lin et al demonstrated the synthesis of calothrixin B 904 from indole 331 and diene 973.88 The Pd(II)-catalyzed tandem cyclization of indole 331 with diene 973 triggered by oxidative C3-alkenylation of indole produced carbazolelactam 974. Removal of the PMB protecting group in 974 yielded 975. Triflation of compound 975 followed by reduction with a palladium catalyzed system [Pd(OAc)2-HCOOH-DPPF] afforded phenanthridine 976. Hydrolysis of 976, followed by Curtis rearrangement produced amine 977. Finally, oxidation of 977 with molecular oxygen under basic conditions resulted in calothrixin B 904 (Scheme 98).
Total Synthesis of Calothrixin B 904 by Lin et al.
Singh et al developed a short and efficient strategy for the synthesis of carbazole derivatives from 2-chloroindole-3-ynones with nitromethanes. The approach was applied for the total synthesis of calothrixin B 904 and staurosporinone 978.23 Initially, the one-pot domino benzannulation reaction between indole-3-ynone 979 and nitroethane 175 catalyzed by Cs2CO3 delivered carbazole 980 in excellent yield. MOM protection of 980 gave 981, which underwent Cadogan reductive insertion of nitro group onto the adjacent phenyl ring to generate indolocarbazole 982. Vilsmeier-Haack reaction of 982 followed by N-MOM protection afforded 984. CAN mediated one-pot oxidation/hydrolysis/quinoline formation of 984 furnished 972, which was converted to calothrixin B 904 by debenzylation (Scheme 99).
Total Synthesis of Calothrixin B 904 and Staurosporinone 978 by Singh et al.
For the synthesis of staurosporinone 978, benzannulation reaction between indole-3-ynone 979 and nitroethane 985 produced carbazole 986. This reaction could be efficiently expanded in gram scale. Trilation of 986 resulted in 987, which underwent Pd-mediated coupling with Zn(CN)2 to form cyanated nitrocarbazole 988. Cadogan reductive cyclization on 988 using monowave furnished pentacyclic indolo-carbazole 989. Raney-Ni reduction of the cyano group in 989 and concomitant lactamization provided 990, which was transformed into staurosporinone 978 by benzyl removal reaction (Scheme 99).
In 2021, Liu et al completed the synthesis of calothrixins A 911 and B 904 based on Baylis-Hillman/6π electrocyclization/ dehydroaromatization sequence of acrylamide-ketone.89 The DBU-catalyzed domino reaction of acrylamide-ketone 991 produced pentacyclic lactam 992. Reduction of this lactam by LiAlH4/AlCl3 gave dihydro-intermediate, which was oxidated to phenanthridine 993 using molecular oxygen. DMP oxidation of 993 in DMSO at elevated temperature furnished calothrixin B 904, which was oxidized to calothrixin A 911 using m-CPBA (Scheme 100).
Total Synthesis of Calothrixins A 911 and B 904 by Liu et al.
Deng et al described the total synthesis of ellipticine 994 and calothrixin B 904 based on an intramolecular Diels-Alder cyclization/dehydro-aromatization tandem.90 Intramolecular Diels-Alder reaction of 995 delivered 996, which was reduced to primary alcohol 997 using BF3.Me2S. N-Boc protection of 997 yielded 998, which was converted to 999 by a sequence of hydrogenation and deprotection. Oxidation of 999 resulted in ellipticine 994 along with 3,4-dihydro ellipticine 1000. The Intramolecular Diels-Alder reaction of 1001 to prepare 1002 was catalyzed by iodine. Reduction of ester in 1002 afforded primary alcohol 1003, which underwent regioselective oxidation using I2/ DMSO to give phenanthridine 1004. IBX oxidation of 1004 yielded 1005, which was oxidized to calothrixin B 904 by H2O2, (PhSe)2 (Scheme 101).
Total Synthesis of Ellipticine 994 and Calothrixin B 904 by Deng et al.
Fox et al reported the total synthesis of indolocarbazole alkaloid staurosporinone 978via a sequential C-H functionalisation strategy.91 The ortho-selective arylation of 1006 by Ph2IOTf 1007 proceeded well under Cu(OTf)2 catalysis to prepare biaryl 1008. Amide 1009 was obtained from 1008 by a sequence of hydrogenolysis and carbamoylation with propionyl chloride. The arylation of 1009 by Ph2IOTf 1007 to synthesize 1010 was achieved by the use of CuI, AgOTf as a catalyst system. Nitration of 1010 afforded 1011, which underwent the copper-catalyzed intramolecular oxidative C–H amination to generate carbazole 1012. Bisbromination of the methyl side chain of 1012 followed by treatment of the resulting dibromide with DMF and K2CO3 provided aldehyde 1013. Reductive amination of 1013 with benzyl amine 1014 gave secondary amine 1015, which was transformed into lactam 1016 by palladium-catalyzed C–H carbonylation. Cadogan cyclization of 1016 furnished 1017, which was converted to staurosporinone 978 by deprotection using TBAI and BCl3 (Scheme 102).
Total Synthesis of Staurosporinone 978 by Fox et al.
Saha et al accomplished the total synthesis of staurosporinone 978 and arcyriaflavin A 958 based on benzannulation of 2-alkenylindoles with 1,3-dicarbonyls.92 Carbazole 1020 was obtained from benzannulation of 2-alkenylindole 1018 with methyl acetoacetate 1019 through a sequence of alkenylation and 6π-electrocyclization. Sulfonylation of 1020 delivered electron-deficient carbazole 1021, which was transformed into lactam 1022 by a sequence of benzylic bromination, nucleophilic substitution of the resulting bromide with ammonia, and intramolecular amidation. Cadogan cyclization of 1022 followed by desulfonation in basic medium furnished staurosporinone 978. Benzannulation of 2-alkenylindole 1018 with 1,3-dicarbonyl 1023 afforded indolocarbazole 1024. Treatment of 1024 with KOH in ethanol under reflux conditions followed reaction of the resulting anhydride with neat ammonium acetate at 140 °C furnished arcyriaflavin A 958 (Scheme 103).
Total Synthesis of Staurosporinone 978 and Arcyriaflavin A 958 by Saha et al.
Men et al developed an efficient approach to access carbazole derivatives by formal [1 + 2 + 3] annulation of oalkenyl arylisocyanides with α, β-unsaturated ketones. The methos was successfully applied to prepare arcyriaflavin A 958 and racemosin B 1025.93 The formal [1 + 2 + 3] annulation between o-alkenyl arylisocyanide 1026 and α,β-unsaturated ketone 1027 provided carbazole 1028 without using a catalyst. Treatment of 1028 with KOH at reflux gave the corresponding anhydride, which was transformed into imide 1029 by reaction with NH4OAc. Cadogan cyclization of 1029 then furnished arcyriaflavin A 958. Similarly, the annulation between o-alkenyl arylisocyanide 1010 and α,β-unsaturated ketone 1031 delivered carbazole 1032, which was transformed into racemosin B 1025 by Cadogan cyclization (Scheme 104).
Total Synthesis of Arcyriaflavin A 958 and Racemosin B 1025 by Men et al.
Kotha et al demonstrated an efficient and short approach for the synthesis of tjipanazoles D 1033 and I 1034 by a shott route from simple starting materials.94 Fischer indolization reaction between phenyl hydrazine 1035 with ketone 1036 provided tetrahydrocarbazole 1037. Regioselective oxidation of 1037 using periodic acid gave 1038. Fischer indolization reaction between phenyl hydrazine 1035 with 1038 furnished tjipanazole D 1033, while the use of phenyl hydrazine 1039 delivered tjipanazole I 1034 (Scheme 105).
Total Synthesis of Tjipanazoles D 1033 and I 1034 by Kotha et al.
Tao et al achieved the total synthesis of dictyodendrins B 1040 and E 1041 in 9 steps and 27% yield and 11 steps and 29% yield, respectively.95 Ullmann coupling of m-dinitrobenzene 1042 and p-iodoanisole 1043 gave diaryl 1044, which underwent aromatic substitution with NaOMe to form 1045. Reduction of nitro 1045 resulted in aniline 1046, which was converted to 1047 by iodonization with ICl. Reaction between o-iodoaniline 1047 and alkyne 1048 produced indole 1049 and 1050 after removal of TES group using TBAF. Alkylation of 1050 with bromide 1051 delivered 1052, which underwent Fridel-Crafts acylation with chloride acid 1053 to yield 1054. Bromination of 1054 provided bromide 1055, which was converted to 1057 by reaction with amine 1056. Benzyl cleavage of 1057 by hydrogenation furnished 1058. Finally, dictyodendrin B 1040 was achieved from 1058 by successive treatment with ClSO2CH2CCl3, BBr3 and TBAI, and Zn dust, NH4OAc. Diazotization of aniline 1046 followed by iodination afforded iodide 1059, which underwent Ullmann coupling with BocNHNH21060 to form 1061. Treatment of compound 1061 and ketone 1062 with PTSA in EtOH resulted in indole 1063, which was converted to 1064 by alkylation with bromide 1051. Bromination of 1064 produced 1065, which was transformed into 1066 by reaction with amine 1056. Benzyl removal of 1066 delivered 1067. Transformation of 1067 to 1068 was similar to the last step of synthesis of dictyodendrin B. Oxidation of 1068 with DDQ completed the total synthesis of dictyodendrin B 1040 (Scheme 106).
Total Synthesis of Dictyodendrins B 1040 and E 1041 by Tao et al.
Pitts et al demonstrated a strategy for the total synthesis of dictyodendrin B 1040 in gram scale.96 Firstly, copper-catalyzed C-H arylation of bromoindole 1069 using diaryliodonium salt 1070 afforded disubstituted indole 1071. Bi(OTf)3−catalyzed Friedel–Crafts acylation of 1071 with 1053 yielded 2,3-disubstituted indole 1072. Borylation at the C7 position of indole 1072, followed by Suzuki coupling with iodide 1043 then provided 1073. N-alkylation of NH-indole 1073 with 4-methoxyphenethyl bromide 1051 gave 1074, which underwent Suzuki–Miyaura coupling with boronic ester 1075 to form 1076. Bromination of 1076 with NBS, followed by treatment of the resulting bromine with NaOMe/CuI delivered 1077. Nitro 1077 was converted to azide 1078 by a sequence of reduction/diazotization/azidation. Thermal decomposition of this azide followed by intramolecular N-H amination then furnished 1079. Finally, dictyodendrin B 1040 was achieved from 1079 by a sequence of selective removal of the tert-butyl ether, sulfonylation, global demethylation, and zinc-mediated cleavage of the sulfonyl protecting group (Scheme 107).
Total Synthesis of Dictyodendrin B 1040 by Pitts et al.
Zhang and Ready accomplished the total synthesis of dictyodendrins F 1080, H 1081, and I 1082.97 Sonogashira coupling of 3-iodoindole 1083 with terminal alkene 1084 produced 3-alkynylindole 1085. Heating 1085 to 50 °C afforded indolyl ketene 1086, which underwent [2 + 2]-cycloaddition reaction with alkyne 1087 to generate cyclobutenone 1088 in moderate yield, this is the key step for the synthesis. Reaction between 1088 and amine 1089 funished 1090, which reacted with 3-pentyl anisolyl ynol ether 1091 to give N-acylated carbazole 1092via a one-pot electrocyclization/acylation sequence. Treatment of 1092 with LiHMDS at −78 °C furnished 1093, which was transformed into dictyodendrin F 1080. The synthesis of dictyodendrins H 1081 and I 1082 from cyclobutenone 1088 followed te same procedure with the employment of amine 1094 and 1095, respectively (Scheme 108).
Total Synthesis of Dictyodendrins F 1080, H 1081, and I 1082 by Zhang and Ready.
Matsuoka et al achieved the total and formal synthesis of dictyodendrins B 1040, E 1041, F 1080, A 1102, C 1103, and D 1104.98 Sonogashira coupling of 1105 with alkyne 473, follow by base-promoted TMS cleavage gave phenyl acetylene 1107. Cadiot–Chodkiewicz coupling of 1107 with bromoalkyne 1108 afforded diyne 1109, which was converted to 1110 by Boc deprotection with TMSOTf and 2,6-lutidine. Azidation of aniline 1110 using tBuONO and TMSN3 afforded azide 1111, which reacted with pyrrole 1112 to yield pyrrolo[2,3-c]carbazole 1113. Base-promoted Boc cleavage of 1113 delivered 1114, which was converted to 1115 by a sequence of bromination with NBS, N-alkylation with bromide 1051, and Suzuki–Miyaura coupling with anisylboronic acid 323. Bromination of 1115 followed by selective debromination of the resuting dibromide afforded monobromide 1116. The Ullmann type reaction of this bromide with NaOMe in the presence of CuI delivered 1117. The total synthesis of dictyodendrin F 1080 was completed through deprotection of 1117 with BBr3 and aerobic oxidation (Scheme 109). Compound 1117 is a known precusor of dictyodendrin C 1103.
Total and Formal Synthesis of Dictyodendrins B 1040, E 1041, F 1080, A 1102, C 1103, and D 1104.
Selective bromination of 1115 resulted in bromide 1118, which was successively treated with MeLi/BuLi and p-anisaldehyde 1119 to form 1120. Conversion of 1120 to 1121 was achieved through a sequence of bromination, oxidation, and Ullmann type coupling. Compound 1121 is a known precusor of dictyodendrins B 1040 and E 1041. The synthesis of 1124 from 1114 was similar to the synthesis of 1117 from 1114 with the use of bromide 1122 instead of 1051. Compound 1124 is a known precusor of dictyodendrin D 1104. Acylation of 1117 with oxalyl chloride followed by treatment with methanol produced 1126. Base-catalyzed hydrolysis of 1126 followed by nucleophilic addition with Grignar reagent 1127 furnished 1128. Methylation of 1128 with TMSCH2N2 followed by hydrogenation provided 1129, a known precusor of dictyodendrin A 1102 (Scheme 109).
In 2022, Munda et al described the total syntheses of xiamycins C-F 1129-1132.99 The syntheses started from tricyclic compound 1133, which was prepared from commercially available abietic acid. Bromination of 1133 resulted in 1134, which underwent nitration with fuming HNO3 to give 1135. Suzuki–Miyaura coupling of 1135 with phenylboronic acid 186 delivered 2-phenylnitrobenzene 1136, which was converted to deoxyxiamycin A methyl ester 1137 by Cadogan's ring closure. Next, compound 1139 was obtained from 1137 by a sequence of N-tosylation and benzylic oxidation using CrO3. Oxidation of ketone 1139 by SeO2 afforded the corresponding enone 1141 (via1140) in 82% yield. Interestingly, bromination of 1141 using NBS with acid catalyst furnished 1142 in excellent yield. Treatment of 1142 with DBU gave 1143, which was oxidized by SeO2 to form 1144. Hydrogenation of 1144 provided 1145, which was reduced to 1146 by NaBH4. Removal of Ts group in 1146 using Mg/MeOH furnished xiamycin D 1130, which was oxidized to xiamycin E 1131 by MnO2. Based-catalyzed hydrolysis of 1131 then produced xiamycin F 1132. Finally, reduction of 1132 using NaBH4 yielded xiamycin C 1129 (Scheme 110).
Total Syntheses of Ciamycins C-F 1129-1132.
A suggested reaction mechanism, that explains the exclusive formation of 1307 from 1306, is outlined at the end of Scheme 110. Initially, protonation of 1307 generates intermediate A, which undergoes a synselective 1,2-migration of the angular methyl group to give intermediate B. Deprotonation of B forms intermediate C, which reacts with NBS to give intermediate D. Ring opening of D followed by syn-selective 1,2-migration of the methyl group then furnished 1307. In control experiments, intermediate C was isolated in 35% yield by treatment of 1307 with AcOH and H2SO4 at room temperature. Bromination of the intermediate C provided 1307 in 84%.
Synthesis of Natural bis Carbazole Alkaloids
In 2014, the Knölker achieved the total synthesis of bismurrayafoline-A 1147, bismurrayafolinol 1148 and chrestifolines B-D 1149-1151.100 The synthesis employed mukonine 109 and murrayafoline-A 110 as the key building blocks. Synthesis of these compound were presented in Scheme 7.
Boc protection of 109 generated 1152, which was reduced to primary alcohol 1153 by LiAlH4. This compound was converted to PNB ether 1154, which contains a good leaving group, before participating Ullmann-type coupling with 109 to give biscarbazole 1155. Reduction of 1155 delivered bismurrayafolinol 1148, which could be oxidized to chrestifoline D 1151. Similarly, Ullmann-type coupling of 1154 with murrayafoline-A 110 produced bismurrayafoline-A 1147. Ussing the same fashion, reaction between 1154 with girinimbine 714 and mahanimbine 715 afforded chrestifoline-B 1149, and chrestifoline-C 1150, respectively (Scheme 111).
Total Synthesis of Bismurrayafoline-A 1147, Bismurrayafolinol 1148 and Chrestifolines B-D 1149-1151.
In 2016, the group accomplished the total synthesis of bismurrayafoline-A 1147, bismurrayafolinol 1148, chrestifolines B-D 1149-1151, murrafoline-E 1156, and murrastifoline-C 1157.15 The synthesis of bismurrayafoline-A 1147, bismurrayafolinol 1148, chrestifolines B-D 1149-1151 was followed their previous report with improvement in reaction yields of several steps.100
The synthesis of murrafoline-E 1156 started from girinimbine 714. Oxidation of the methyl side chain in 714 gave murrayacine 593, which was then protected as N-Boc 1158. Reduction of 1158 by NaBH4 afforded primary alcohol 1159, which was converted to PNB ether 1160. Ullmann-type coupling of 1160 with 110 furnished murrafoline-E 1156. The same procedure was applied to prepare murrastifoline-C 1157 from murrayacinine 593 (Scheme 112).
Total Synthesis of Bismurrayafoline-A 1147, Bismurrayafolinol 1148, Chrestifolines B-D 1149-1151, Murrafoline-E 1156, and Murrastifoline-C 1157.
Banerjee et al completed the synthesis of sorazolon E2 1164 from sorazolon E 173.83 The synthesis of Sorazolons E was presented in Scheme 49. Dimerization of sorazolon E in the presence of CuSO4/Al2O3 under an O2 atmosphere at 140 °C furnished (±)-sorazolon E2 (Scheme 113).
Synthesis of (±)-Sorazolon E2.
Conclusion
In this review article, a great number of studies on total and formal synthesis of natural carbazole alkaloids dating back 2014 have been summarized. The construction of carbazole skeleton by oxidative cyclization of diaryl amines still attracted great attention of chemists. More importantly, different strategies for the synthesis of natural carbazole alkaloids have been developed. A wide range of complex carbazole alkaloids such as fused carbazole alkaloid, dimeric carbazole alkaloid, and indolocarbazole carbazole alkaloid has been synthesized efficiently. In several works, the total synthesis required more than 15 steps with many chiral centres. Undoubtedly, more and more innovative synthetic strategies for the synthesis of natural carbazole alkaloids will be appeared in the future due to challenging structure and interesting bioactivities of this class of alkaloid. Development of greener synthetic methodologies and cost-effective catalysts will be examined. Exploration of enzymatic routes for carbazole alkaloid biosynthesis might be another direction. Application of established methods for the synthesis of medically important carbazole alkaloids might be another direction of carbazole chemistry.
Footnotes
Abbreviations
Author contributions
Vo Cong Dung collected materials, Nguyen Thi Chung prepared draft, and Dau Xuan Duc wrote the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
SchmidtAWReddyKRKnölkerH-J. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem Rev. 2012;112(6):3193–3328. Doi: https://doi.org/10.1021/cr200447s
3.
GregerH. Phytocarbazoles: alkaloids with great structural diversity and pronounced biological activities. Phytochem Rev. 2017;16:1095–1153. Doi: https://doi.org/10.1007/s11101-017-9521-5
ChamberGESayanAEBrownRCD. The synthesis of biologically active indolocarbazole natural products. Nat Prod Rep. 2021;38:1794–1820. DOI: https://doi.org/10.1039/d0np00096e
6.
KnöllerHJReddyKR. Isolation and synthesis of biologically active carbazole alkaloids. Chem Rev. 2002;102(11):4303–4428. Doi: https://doi.org/10.1021/cr020059j
7.
HiedaF. Total syntheses of multi-substituted carbazole alkaloids and phenolic related compounds, and evaluation of their antioxidant activities. Yakugaky Zasshi. 2017;137(10):1255–1263. Doi: https://doi.org/10.1248/yakushi.17-00142
8.
HesseRKataevaOSchmidtAWKnölkerH-J. Synthesis of prenyl- and geranyl-substituted carbazole alkaloids by DIBAL-H promoted reductive pyran ring opening of dialkylpyrano[3,2-a]carbazoles. Chem Eur J. 2014;20(31):9504–9509. Doi: https://doi.org/10.1002/chem.201403645
9.
HesseRJägerASchmidtAWKnölkerH-J. Palladium(ii)-catalysed total synthesis of naturally occurring pyrano[3,2-a]carbazole and pyrano[2,3-b]carbazole alkaloid. Org Biomol Chem. 2014;12:3866–3876. Doi; 10.1039/C4OB00367E
10.
HesseRKrahlMPJägerAKataevaOSchmidtAWKnölkerH-J. Palladium(II)-catalyzed synthesis of the formylcarbazole alkaloids murrayaline A–C, 7-methoxymukonal, and 7-methoxy-O-methylmukonal. Eur J Org Chem. 2014;19:4020–4028. Doi: https://doi.org/10.1002/ejoc.201402201
11.
SchusterCBörgerCJulich-GrunerKK, et al.Synthesis of 2-hydroxy-7-methylcarbazole, glycozolicine, mukoline, mukolidine, sansoakamine, clausine-H, and clausine-K and structural revision of clausine-TY. Eur J Org Chem. 2014;22:4741–4752. Doi: https://doi.org/10.1002/ejoc.201402495
12.
HesseRSchmidtAWKnölkerH-J. Total synthesis of glycomaurrol and eustifoline-C by DIBAL-H promoted reductive ring opening of pyrano[2,3-c]carbazoles. Tetrahedron. 2015;71(21):3485–3490. Doi: https://doi.org/10.1016/j.tet.2015.03.064
13.
KutzSKSchmidtAWKnölkerH-J. Palladium-catalyzed synthesis of pyrayaquinones, murrayaquinones, and murrayafoline-B. Synthesis (Mass). 2016;48(02):275–292. Doi: https://doi.org/10.1055/s-0036-1588322
14.
SchusterCRönnefahrtMJulich-GrunerKKJägerASchmidtAWKnölkerH-J. Synthesis of the pyrano[3,2-a]carbazole alkaloids koenine, koenimbine, koenigine, koenigicine, and structural reassignment of mukonicine. Synthesis (Mass). 2016;48(01):150–160. Doi: https://doi.org/10.1055/s-0035-1560359
15.
KutzSKBörgerCSchmidtAWKnölkerH-J. Synthesis of methylene-bridged biscarbazole alkaloids by using an ullmann-type coupling: first total synthesis of MurrastifolineC and murrafoline-E. Chem Eur J. 2016;22(7):2487–2500. Doi: https://doi.org/10.1002/chem.201504680
16.
BrüttingCHesseRJägerAKataevaOSchmidtAWKnölkerH-J. Synthesis of glycoborine, glybomine A and B, the phytoalexin carbalexin A and the b-adrenoreceptor antagonists carazolol and carvedilol. Chem Eur J. 2016;22(47):16897–16911. Doi: https://doi.org/10.1002/chem.201604002
17.
BrüttingCKataevaOSchmidtAWKnölkerH-J. First total synthesis of the cytotoxic carbazole alkaloid excavatine-A and regioselective annulation to pyrano[2,3- a]carbazoles and [1,4]oxazepino[2,3,4-jk]carbazoles. Eur J Org Chem. 2017;22:3288–3300. Doi: https://doi.org/10.1002/ejoc.201700515
18.
KishontiAJägerAKnölkerH-J. Synthesis of clausenal, 1,5-dimethoxycarbazole-3-carbaldehyde and 2,5-dimethoxycarbazole-3-carbaldehyde. Eur J Org Chem. 2020;34:5572–5579. Doi: https://doi.org/10.1002/ejoc.202000917
19.
MaDDaiJQiuYFuCMaS. Gram scale synthesis of 7-methoxyO-methylmukonal, clausine-O, clausine-K, clausine-H, 7-methoxymukonal, and methyl 2-hydroxy-7-methoxy-9H-carbazole- 3-carboxylate. Org Chem Front. 2014;1:782–791. Doi: https://doi.org/10.1039/c4qo00163j
20.
Taufiq-YapYHPehTHEeGCL, et al.A new cytotoxic carbazole alkaloid from Clausena excavata. Nat Prod Res. 2007;21(9):810–813. Doi: 0.1080/14786410701258875
21.
MarkadSBArgadeNP. Diversity oriented convergent access for collective total synthesis of bioactive multifunctional carbazole alkaloids: synthesis of carbazomycin A, carbazomycin B, hyellazole, chlorohyellazole, and clausenaline D. Org Lett. 2014;16(20):5470–5473. Doi: https://doi.org/10.1021/ol502721r. Doi: 10.1021/ol502721r
22.
PolleyAVaralaxmiKNandiAJanaR. Divergent total synthesis of (-)-mahanine and other carbazole alkaloids. Asian J Org Chem. 2021;10:1207–1215. Doi: https://doi.org/10.1002/ajoc.202100176
23.
SinghSSamineniRPabbarajaSMehtaG. A general carbazole synthesis via stitching of indole-ynones with nitromethanes: application to total synthesis of carbazomycin A, calothrixin B, and staurosporinone. Org Lett. 2019;21(9):3372–3376. Doi: https://doi.org/10.1021/acs.orglett.9b01111
24.
RoyJMalD. Total synthesis of carbazole alkaloids – ekeberginine, harmandianamine A, and furanoclausamine B. Eur J Org Chem. 2014;9:1873–1881. Doi: 0.1002/ejoc.201301652
HumneVDangatYVankabKLokhandeP. Iodine-catalyzed aromatization of tetrahydrocarbazoles and its utility in the synthesis of glycozoline and murrayafoline A: a combined experimental and computational investigation. Org Biomol Chem. 2014;12:4832–4836. Doi: https://doi.org/10.1039/c4ob00635f
27.
NaykodeMSTambeSDLokhandePD. I2-Catalyzed total synthesis of collective 7-oxygenated carbazole alkaloids: clauszoline-K, -L, Clausine-M, -N and formal synthesis of siamenol. ChemistrySelect. 2017;2(1):567–570. Doi: https://doi.org/10.1002/slct.201601759
28.
DharaKMandalTDasJDashJ. Synthesis of carbazole alkaloids by ring-closing metathesis and ring rearrangement–aromatization. Angew Chem Int Ed. 2015;54(52):15831–15835. Doi: https://doi.org/10.1002/anie.201508746
29.
BanerjeeASahuSMajiMS. Benzannulation of 2-alkenylindoles using aldehydes by sequential triple-relay catalysis: a route to carbazoles and carbazole alkaloids. Adv Synth Catal. 2017;359(11):1860–1866. Doi: https://doi.org/10.1002/adsc.201700092
30.
ChakrabortySSahaC. Total synthesis of marine alkaloid hyellazole and its derivatives. Eur J Org Chem. 2018;17:2013–2021. Doi: https://doi.org/10.1002/ejoc.201800187
31.
AlimiIRemyRBochetCG. Photochemical C-H activation: generation of indole and carbazole libraries, and first total synthesis of clausenawalline D. Eur J Org Chem. 2017;22:3197–3210. Doi: https://doi.org/10.1002/ejoc.201700300
32.
QiuYZhouJFuCShengmingM. A general diversified synthesis of carbazoles and the first synthesis of karapinchamine A. Chem Eur J. 2014;20(45):14589–14593. Doi: https://doi.org/10.1002/chem.201404769
ElumalaiVGambarottiCBjørsvikHCarbazomycinG. Method development and total synthesis. Eur J Org Chem. 2018;17:1980–1992. Doi: https://doi.org/10.1002/ejoc.201800359
35.
KumarKAKannaboinaPJaladankiCKBharatamPVDasP. Copper-Catalyzed N-arylation of tautomerizable heterocycles with boronic acids and its application to synthesis of oxygenated carbazoles. ChemistrySelect. 2016;1(3):601–607. Doi: https://doi.org/10.1002/slct.201600147
AnYWangYHuX. Total synthesis of carbazomycin G by a thermal ring expansion/self-redox reaction cascade. Eur J Org Chem. 2014;17:3715–3718. Doi: https://doi.org/10.1002/ejoc.201402065
38.
LiuYGuoYJiFGaoDSongCChangJ. Divergent syntheses of carbazole alkaloids clausenapin, indizoline, claulansine M, and clausenaline D. J Org Chem. 2016;81(10):4310–4315. Doi: https://doi.org/10.1021/acs.joc.6b00729
Hernández-BenitezRIZárate-ZárateDDelgadoFTamariJ. Palladium-Catalyzed synthesis of diarylamines and 1- and 2-oxygenated carbazoles: total syntheses of natural alkaloids clauraila A, clausenal, clausine P, and 7-methoxy-O-methylmukonal. Synthesis (Mass). 2017;49(18):4357–4371. Doi: https://doi.org/10.1055/s-0036-1588467
41.
RasheedSRaoDNReddyKRAravindaSVishwakarmaRADasP. C–N bond formation via Cu-catalyzed crosscoupling with boronic acids leading to methyl carbazole-3-carboxylate: synthesis of carbazole alkaloids. RSC Adv. 2014;4:4960–4969. Doi: https://doi.org/10.1039/c3ra44903c
42.
KaliyaperumalSABanerjeeSKumarSUK. Palladium mediated intramolecular multiple C–X/C–H cross coupling and C–H activation: synthesis of carbazole alkaloids calothrixin B and murrayaquinone A. Org Biomol Chem. 2014;12:6105–6113. Doi: https://doi.org/10.1039/ c4ob00493k
43.
GooDYWooSK. One-pot synthesis of carbazoles via tandem C-C cross-coupling and reductive amination. Org Biomol Chem. 2016;14:122–130. Doi: https://doi.org/10.1039/C5OB01952D
KaranGSahuSMajiMS. A one-pot ‘‘back-to-front’’ approach for the synthesis of benzene ring substituted indoles using allylboronic acids. Chem Commun. 2021;57:5274–5277. Doi: https://doi.org/10.1039/D1CC01512E
46.
PolleyAVaralaxmiKJanaR. Palladium-Catalyzed ortho C-H arylation of aniline carbamates with diazonium salts under mild conditions: expedient synthesis of carbazole alkaloids. ACS Omega. 2018;3(10):14503–14516. Doi: https://doi.org/10.1021/acsomega.8b02009
47.
MominAAUrmodeTDBhosaleSM. Kusurkar RSA short synthesis of carbazole alkaloids - ekeberginine, murrayaquinone A and glycozoline. Synth Commun. 2016;46(15):1292–1298. Doi: https://doi.org/10.1080/00397911.2016.1192652
48.
NishiyamaTMatsuokaAHondaRKitamuraTHataeNChoshiT. Total synthesis of carbazole alkaloid clausamine E. Tetrahedron. 2020;76(15):13110. Doi: https://doi.org/10.1016/j.tet.2020.131110
WuSHaradaSMorikawaTNishidaA. Enantioselective total synthesis of a natural hydrocarbazolone alkaloid, identification of its stereochemistry, and revision of its spectroscopic data. Tetrahedron: Asymmetry. 2017;28(8):1083–1088. Doi: https://doi.org/10.1016/j.tetasy.2017.07.005
51.
MarkadSBArgadeNP. Biomimetic collective total synthesis of bioactive carbazole alkaloids indizoline, mafaicheenamine A, claulamine A, claulansine A and the proposed claulamine E. J Org Chem. 2016;81(12):5222–5227. Doi: https://doi.org/10.1021/acs.joc.6b00702
52.
NishiyamaTSatsukiNHibinoS, et al.Total synthesis of carbazole-1,4-quinone alkaloid koeniginequinones A and B based on a one-pot cyclocarbonylation procedure from 2-alkenyl-3-iodoiodoindole. Heterocycles. 2016;93(1):84–100. Doi: https://doi.org/10.3987/COM-15-S(T)5
53.
GaikwadMVKambleRDHeseSV, et al.A short synthesis of carbazole alkaloids murrayanine and mukonine. Chem Methodol. 2021;5(4):341–347. Doi: https://doi.org/10.22034/chemm.2021.131552
54.
PointsGLBeaudryCM. Regioselective synthesis of substituted carbazoles, bicarbazoles, and clausine C. Org Lett. 2021;23(17):6882–6885. Doi: https://doi.org/10.1021/acs.orglett.1c02449
PanXDongBWuYGaoBSongC. Synthesis of functionalized 4-hydroxy carbazoles and carbazole alkaloids via ring expansion of indole cyclopentanone. J Org Chem. 2024;89(12):8845–8850. Doi: https://doi.org/10.1021/acs.joc.4c00730
58.
BanerjeeASahaSMajiMS. Cascade benzannulation approach for the syntheses of lipocarbazoles, carbazomycins, and related alkaloids. J Org Chem. 2022;87(6):4343–4359. Doi: https://doi.org/10.1021/acs.joc.2c00042
59.
BommanaboinaBRoyDBaireB. Integrated synthesis of 3,4-carbazoquinone alkaloids N-me-carbazoquinocin A, B and D–F. RSC Adv. 2024;14:27372–27384. Doi: https://doi.org/10.1039/d4ra05347h
60.
NishiyamaTKiharaYTakeuchiN, et al.Total syntheses of carbazole alkaloid mukoenine A and pyrano[3,2-a] carbazole alkaloid girinimbine. Tetrahedron. 2022;120(13):132895. Doi: https://doi.org/10.1016/j.tet.2022.132895
61.
GassnerCHesseRSchmidtAWKnölkerH-J. Total synthesis of the cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids derived from 2-hydroxy-6-methylcarbazole. Org Biomol Chem. 2014;12:6490–6499. Doi: https://doi.org/10.1039/c4ob01151a
62.
Julich-GrunerKKKataevaOSchmidtAWKnölkerH-J. Total synthesis of 7- and 8-oxygenated pyrano[3,2-a]carbazole and pyrano[2,3-a]carbazole alkaloids via boronic acid-catalyzed annulation of the pyran ring. Chem Eur J. 2014;20(28):8536–8540. Doi: https://doi.org/10.1002/chem.201403143
63.
SchusterCJulich-GrunerKKSchnitzlerH, et al.Total synthesis of murrayamine-E, murrayamine-I, and murrayamine-K. J Org Chem. 2015;80(11):5666–5673. Doi: https://doi.org/10.1021/acs.joc.5b00630
64.
BrüttingVSchmidtAWKataevaOKnölkerH-J. First total synthesis of 7-isovaleryloxy-8-methoxygirinimbine. Synthesis (Mass). 2018;50(13):2516–2522. Doi: https://doi.org/10.1055/s-0037-1609717
65.
LösleVKataevaOKnölkerH-J. First total synthesis and investigation of the x-ray crystal structure of the pyrano[3,2-a]carbazole alkaloid clausenalansine A. Synthesis (Mass). 2020;52(02):359–364. Doi: https://doi.org/10.1055/s-0040-1706551
66.
HouSLiuYKongYBrownML. Total synthesis of 7-hydroxymurrayazolinine, murrayamine D, and mahanine via m-nitro group activated pyran annulation. Org Lett. 2015;17(10):2298–2301. Doi: https://doi.org/10.1021/acs.orglett.5b00422
67.
DaiJMaDFuCMaS. Gram scale total synthesis of 2-hydroxy-3-methylcarbazole, pyrano[3,2-a]carbazole and prenylcarbazole alkaloids. Eur J Org Chem. 2015;25:5655–5662. Doi: https://doi.org/10.1002/ejoc.201500783
68.
DetheDHDasSDherangeBDMahapatraS. Enantiospecific total syntheses and assignment of absolute configuration of cannabinol-skeletal carbazole alkaloids murrayamines-O and -P. Chem Eur J. 2015;21(23):8347–8350. Doi: 0.1002/chem.201406434
69.
ReddKRReddyASDhakedDK, et al.Palladium-catalyzed arylation of 2H-chromene: a new entry to pyrano[2,3-c]carbazoles. Org Biomol Chem. 2015;13:9285–9293. Doi: https://doi.org/10.1039/C5OB01295C
70.
AbbasaYManshaaMUllahN. The first total synthesis of potent antitumoral (±)-mafaicheenamine A, unnatural 6-fluoromafaicheenamine A and expedient synthesis of clausine E. RSC Adv. 2016;6:26104–26110. Doi: https://doi.org/10.1039/C6RA03242G
71.
WangTHoyeTR. Hexadehydro-Diels-Alder (HDDA)-enabled carbazolyne chemistry: single step, de Novo construction of the pyranocarbazole core of alkaloids of the Murraya koenigii (curry tree) family. J Am Chem Soc. 2016;138(42):13870–13873. Doi: https://doi.org/10.1021/jacs.6b09628
72.
ZangY-DLiC-JSongX-Y, et al.Total synthesis and neuroprotective effect of O-methylmurrayamine A and 7-methoxymurrayacine. J Asian Nat Prod Res. 2017;19(6):623–629. Doi: https://doi.org/10.1080/10286020.2017.1327952
73.
LuSShangWYangPWangTYangJWangY. Total synthesis of natural pyrano[3,2-a]carbazole alkaloids. Tetrahedron. 2023;149:133743. Doi: https://doi.org/10.1016/j.tet.2023.133743
74.
DangarSRoyTNoskarSBisaiA. Total synthesis of bicyclomahanimbine by cu(II)- promoted photoredox process. RSC Adv. 2024;14:30110–30115. Doi: https://doi.org/10.1039/D4RA05863A
75.
SpindlerBBKataevaOKnölkerH-J. Enantioselective total synthesis and assignment of the absolute configuration of the furo[3,2-a]carbazole alkaloid furoclausine-B. J Org Chem. 2018;83(24):15136–15143. Doi: https://doi.org/10.1021/acs.joc.8b02426
76.
Martín-TorresIOgallaGYangJ-MRinaldiAEchavarrenAM. Enantioselective alkoxycyclization of 1,6-enynes with gold(I)- cavitands: total synthesis of mafaicheenamine C. Angew Chem Int Ed. 2021;60(17):9339–9344. Doi: https://doi.org/10.1002/anie.202017035
77.
MalDRoyJ. A regioselective facile synthesis of furo[3,4-b]- carbazolones: application to the total synthesis of mafaicheenamine E and claulansine. Org Biomol Chem. 2015;1:6344–6352. Doi: https://doi.org/10.1039/C5OB00575B
78.
GoetzAESilbersteinALCorselloMAGargNK. Concise enantiospecific total synthesis of tubingensin A. J Am Chem Soc. 2014;136(8):3036–3039. Doi: https://doi.org/10.1021/ja501142e
79.
GuoYHuangHZhangT, et al.First total synthesis of carbazole alkaloids lansine F and clauemarazole D. Asian J Org Chem. 2016;5(10):1269–1272. Doi: https://doi.org/10.1002/ajoc.201600311
80.
KoberUKnölkerH-J. Palladium-Catalyzed approach to malasseziazole A and first total synthesis of malasseziazole C. Synlett. 2015;26(11):1549–1552. Doi: https://doi.org/10.1055/s-0034-1380713
81.
DongJZhangDMenYZhangXHuZXuX. [1 + 2 + 3] annulation as a general access to indolo[3,2-b]carbazoles: synthesis of malasseziazole C. Org Lett. 2019;21(1):166–169. Doi: https://doi.org/10.1021/acs.orglett.8b03646
82.
SaravananVRamalingamBMMohanakrishnanAK. Total synthesis of calothrixin B and its analogs. Eur J Org Chem. 2014;6:1266–1279. Doi: https://doi.org/10.1002/ejoc.201301524
83.
RamkumarNNagarajanR. Total synthesis of ellipticine quinones, olivacine, and calothrixin B. J Org Chem. 2014;79(2):736–741. Doi: https://doi.org/10.1021/jo402593w
84.
RamkumarNNagarajanR. Formal total synthesis of calothrixin B and its N-benzyl analogues. RSC Adv. 2015;5:87838–87840. Doi: https://doi.org/10.1039/C5RA18120H
85.
RamkumarNNagarajanR. Total synthesis of calothrixin B via sequential sonogashira coupling/copper-catalyzed oxidative cyclization. Org Biomol Chem. 2015;13:11046–11051. Doi: https://doi.org/10.1039/C5OB01766A
86.
Mori-QuirozLMDekarskeMMPrinkkiABCliftMD. Exploiting alkylquinone tautomerization for the total synthesis of calothrixin A and B. J Org Chem. 2017;82(23):12257–12266. Doi: https://doi.org/10.1021/acs.joc.7b02101
87.
LeeSKimK-HCheonC-H. Total syntheses of arcyriaflavin A and calothrixin B using 2,2′- bisindole-3-acetic acid derivative as a common intermediate. Org Lett. 2017;19(11):2785–2788. Doi: https://doi.org/10.1021/acs.orglett.7b00687
88.
LinKJianYZhaoPZhaoCPanWLiuS. A rapid construction of specific quino[4,3-b] carbazolone system and its application for synthesis of calothrixin B. Org Chem Front. 2018;5:590–594. Doi: https://doi.org/10.1039/C7QO00864C
89.
LiuYXuMXieKLiuS. Total synthesis of calothrixin B via an intramolecular BaylisHillman cyclization/6π electrocyclization/dehydroaromatization sequence and a specific oxidative quinone formation. Adv Synth Catal. 2021;363(3):737–741. Doi: https://doi.org/10.1002/adsc.202001231
90.
DengCLiuYXuMXieKLiuS. Exploiting an intramolecular Diels–Alder cyclization/dehydro-aromatization sequence for the total syntheses of ellipticines and calothrixin B. Org Biomol Chem. 2021;19:1395–1403. Doi: https://doi.org/10.1039/d0ob02527e
91.
FoxJCGilliganREPittsAKBennettHRGauntMJ. The total synthesis of K-252c (staurosporinone) via a sequential C–H functionalisation strategy. Chem Sci. 2016;7:2706–2710. Doi: https://doi.org/10.1039/C5SC04399A
92.
SahaSBanerjeeAMajiMS. Brønsted acid catalyzed one-pot benzannulation of 2-alkenylindoles under aerial oxidation: a route to carbazoles and indolo[2,3-a]carbazole alkaloids. Org Lett. 2018;20(21):6920–6924. Doi: https://doi.org/10.1021/acs.orglett.8b03063
93.
MenYHuZDongJXuXTangB. Formal [1 + 2 + 3] annulation: domino access to carbazoles and indolocarbazole alkaloids. Org Lett. 2018;20(17):5348–5352. Doi: https://doi.org/10.1021/acs.orglett.8b02266
94.
KothaSSaifuddinMAswarVR. Diversity-Oriented approach to indolocarbazoles via fischer indolization and olefin metathesis: total synthesis of tjipanazole D and I. Org Biomol Chem. 2016;14:9868–9873. Doi: https://doi.org/10.1039/C6OB01679K
95.
TaoPLiangJJiaY. Total synthesis of dictyodendrins B and E, and formal synthesis of dictyodendrin C. Eur J Org Chem. 2014;26:5735–5748. Doi: https://doi.org/10.1002/ejoc.201402672
96.
PittsAKHaraOSnellFGauntRHJM. A concise and scalable strategy for the total synthesis of dictyodendrin B based on sequential CÿH functionalization. Angew Chem Int Ed. 2015;54(18):5541–5545. Doi: https://doi.org/10.1002/anie.201500067
97.
ZhangWReddyJM. A concise total synthesis of dictyodendrins F, H, and I using aryl ynol ethers as key building blocks. J Am Chem Soc. 2016;138(33):10684–10692. Doi: https://doi.org/10.1021/jacs.6b06460
MundaMNandiRGavitVRKunduSNiyogiSBisaiA. Total syntheses of naturally occurring antiviral indolosesquiterpene alkaloids, xiamycins C–F via Csp3–H functionalization. Chem Sci. 2022;13:11666–11671. Doi: https://doi.org/10.1039/d2sc03479d
100.
BörgerCSchmidtAWKnölkerH-J. First total syntheses of chrestifoline-B and (±)-chrestifoline-C, and improved synthetic routes to bismurrayafoline-A, bismurrayafolinol and chrestifoline-D. Org Biomol Chem. 2014;12:3831–3835. Doi: https://doi.org/10.1039/C4OB00609G