Introduction: 3β-Angeloyloxy-8-oxo-eremophila-6,9-dien-12-oic acid (1) is a natural product isolated from Cacalia (Asteraceae) species. While this compound has been reported to be moderately antibacterial, its absolute configuration at C-11 remains unassigned. More importantly, the absolute configuration at C-11 has only been assigned for two eremophilanoids bearing carboxylic-acid-containing alkyl chains with stereogenic centers. Herein, we describe the preparation of 1 from furanoeremophilane 3 and the assignment of its absolute configuration using vibrational circular dichroism (VCD) spectroscopy. Methods: 3β-Angeloyloxy-6β-hydroxyfuranoeremophil-1(10)-ene (3) was treated with a continuous flux of oxygen while being irradiated with a fluorescent lamp to afford butenolactone 2, which was then subjected to alkaline hydrolysis to yield 1 and 5. The absolute configuration of 1 was assigned using VCD spectroscopy, which involved comparing experimental and density functional theory (DFT) (B3LYP/DGDZVP) calculated spectra of the 3S,4R,5R,11R- and 3S,4R,5R,11S-epimers of methyl ester 4. Results: Butenolactone 2 was obtained by the oxidation of 3, with subsequent hydrolysis affording 1 in moderate yield. Methyl ester 4 was obtained via the Steglich esterification of 1. The 11R and 11S-epimers of 4 were conformationally analyses. The selected conformers were submitted to complete geometry optimization and IR and VCD frequencies calculation using DFT to afford 9 and 8 conformers, contributing with more than 1% of the conformational population, for 11R and 11S, respectively, which were used to generate the calculated IR and VCD spectra. The experimental data were best fitted to the calculated results for the 11R-epimer. Conclusion: 3β-Angeloyloxy-8-oxo-eremophila-6,9-dien-12-oic acid (1) was prepared from 3β-angeloyloxy-6β-hydroxyfuranoeremophil-1(10)-ene (3) in the same configuration as the natural product, as evidenced by comparing the NMR data with those previously reported. A comparison of the calculated IR and VCD spectra of the 11R- and 11S-4 epimers and the experimental spectrum of (+)-4 enabled the 3S,4R,5R,11R absolute configuration to be assigned to natural product 1 isolated from the Cacalia species.
3β-Angeloyloxy-8-oxo-eremophila-6,9-dien-12-oic acid (1) (Figure 1) was first isolated from the methanolic extracts of Cacalia roborowskii roots,1 and some years later, from C. ainsliaeflora was reported to be moderate antibacterial activity against S. aureus, E. coli, and B. subtilis.2,3
Formulas of Eremophilanoids 1-5.
The carboxylic-acid moieties in eremophilanes such as 1 can reportedly be formed from the corresponding butenolactone,4 which, in turn, can be obtained under mild oxidizing conditions.4–6 However, the oxidation of a furoeremophilane to the corresponding lactone by exposure to air and light6–8 is time-consuming and proceeds in low yields. Attempts to establish relationships to other natural products have led to studies into photochemical oxidation with singlet oxygen.9–12
Although the enantiomeric eremophilane series has been described,4 their natural occurrence is limited to a few genera (eg, Alpinia, Cupressus), and no evidence for the existence of these eremophilanes in Cacalia and Senecio species has been reported.13 A few years ago, we used vibrational circular dichroism (VCD) spectroscopy to determine the 4S,5R absolute configuration of the eremophilanolids from Senecio toluccanus.14
Importantly, the absolute configuration at C-11 in 1 was not reported in previous studies on its isolation and biological evaluation,1–3 presumably because the C7–C11–C12 bonds are conformationally free; consequently, deducing the spatial arrangements of these groups through NMR experiments, such as 1D- or 2D-NOESY, is difficult. To our knowledge, the absolute configuration at C-11 has only been assigned twice for this type of eremophilanoid using X-ray diffraction-based anomalous dispersion analysis.15,16 It is worth mentioning that H-11 in the 11R epimer was observed to resonate at a higher frequency (δ 3.70) than that in the 11S epimer (δ 3.59) by 1H NMR spectroscopy in these two cases.15
We previously reported the first isolation of the eremophilanolide 3β-angeloyloxy-6β-hydroxyeremophila-1(10),7(11)-dien-8β−12-olide (2) (Figure 1) as a minor product, along with the 3β-angeloyloxy-6β-hydroxyfuranoeremophil-1(10)-ene furanoeremophilanoid (3) (Figure 1) as the main metabolite of Senecio sinuatus roots.17
We now describe the preparation of 1 via the hydrolysis of 2, which, in turn, was obtained by the oxidation of 3 with singlet oxygen. We also describe, using vibrational circular dichroism spectroscopy, the 11R absolute configuration assignment to 1, which involved calculating IR and VCD spectra for the epimeric methyl 3S,4R,5R,11R- and 3S,4R,5R,11S-angeloyloxy-8-oxo-eremophila-6,9-dien-12-oates (4) (Figure 1) and comparing the results obtained with the experimentally acquired spectra of (+)-4.
Methods
General Experimental Procedures
Merck silica gel 60 (200-400 mesh, ASTM) was used for column chromatography. IR spectra were acquired using a PerkinElmer 1600 FT-IR series instrument equipped with an ATR PRO450-S accessory. NMR spectra were acquired in CDCl3 with TMS as an internal reference using a Varian System 500 spectrometer. HRMS data were obtained in electron-impact mode on a Jeol GCMateII spectrometer. Microwave-assisted reactions were carried out using a CEM Discover equipment.
VCD spectra were acquired on a dual PEM BioTools ChiralIR2X FT-VCD instrument at a resolution of 4 cm−1. Ester 4 was dissolved in CDCl3 (100% D atoms) and placed in a BaF2 cell with a path length of 101 μm, and five 1-h-long data blocks were added. The baseline was corrected by subtracting solvent spectra obtained under identical conditions.
Compounds
3β-Angeloyloxy-6β-hydroxyfuranoeremophil-1(10)-ene (3) was obtained in a previous study.17
The transformation of 3 into 1 and then 4 was repeated several times, with typical procedures described below.
A solution of 3β-angeloyloxy-6β-hydroxyfuranoeremophil-1(10)-ene (3) (200 mg, 0.6 mmol) and methylene blue (20 mg, 0.06 mol) in CH2Cl2 (250 mL) was stirred and irradiated with a 60-W fluorescent lamp for 20 min under a continuous flow of oxygen, which was generated in situ by reacting H2O2 (30%) with 10 M aqueous KI.11,12 The solvent was evaporated under reduced pressure, and the residue was separated by chromatography using hexane/EtOAc as the eluent. Fractions that eluted with 7:3 hexane/EtOAc were re-chromatographed to yield 2 (24.8 mg, 23.6%) as a clear yellow oil. The NMR data provided in the Supplemental Material are essentially the same as those reported in literature17 (Table S2).
3β-Angeloyloxy-6β-hydroxyeremophil-1(10)-en-8β−12-olide (2) (65.3 mg, 0.2 mmol) was dissolved in MeOH (5 ml) and KOH (84.5 mg, 1.51 mmol) was added. The reaction mixture was stirred and subjected to 60-W microwave irradiation for 90 s at 60 °C. The crude reaction mixture was acidified with 5% HCl (0.5 mL) and extracted with EtOAc (15 mL × 3). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was subjected to chromatography using CH2Cl2/MeOH as the eluent to afford 1 (23.0 mg, 35%) as a clear yellow oil.
+ 12.4° (CHCl3, c 0.36), lit.1 [α]D + 38.5° (CHCl3, ca 1.2), Note that the concentration used in reference 1 is described as “ca”. The NMR data for 1, listed in Table S2 in the Supplemental Material, are essentially the same as those reported previously.1
A solution of 3β-angeloyloxy-8-oxo-eremophila-6,9-dien-12-oic acid (1) (58.6 mg, 0.169 mmol) in 7:3 CH2Cl2/MeOH (2 mL) was stirred while cooled in an ice bath. DMAP (1.8 mg, 0.014 mmol) was added, after which DCC (54 mg, 0.203 mmol) was slowly added.18 The reaction mixture was left at room temperature for 3 h, after which the solvent was evaporated under reduced pressure, and the residue was separated by chromatography using hexane/EtOAc as the eluate to afford 4 (21.0 mg, 34.4%) as a clear oil. The NMR data are similar to those described for 1 with the exception of the additional signals for the methoxy group at δ 3.67 and 52.0 in the 1H and 13C spectra, respectively. + 38.1 (c 0.32, CHCl3); IR (cm−1) 2973, 2947, 1737, 1715, 1666, 1636, 1455, 1385, 1232, 1158, 977, 919; HREIMS m/z 360.1935 (calcd. for C21H28O5+, 360.1931); 1H NMR (CDCl3) δ 6.88 (bs, 1H, H-6), 6.17 (d, 1H, J = 1.2 Hz, H-9), 6.13 (qq, 1H, J = 7.1, 1.3 Hz, H-3’), 5.15 (dd, 1H, J = 5.6, 2.9 Hz, H-3), 3.76 (q, 1H, J = 7.1 Hz, H-11), 3.67 (s, 3H, OMe), 2.71 (dddd, 1H, J = 14.5, 13.2, 4.7, 1.5 Hz, H-1β) 2.31 (ddd, 1H, J = 13.3, 4.4, 2.4 Hz, H-1α) 2.21 (dddd, J = 14.4, 4.9, 2.5, 2.5 Hz, H-2β) 2.05 (dq, 3H, J = 7.2, 1.5 Hz, Me-4’) 1.97 (quint, 3H, J = 1.5 Hz, Me-5’) 1.79 (dq, 1H, J = 7.1, 3.4 Hz, H-4), 1.70 (dddd, 1H, J = 14.3, 14.3, 4.5, 3.1 Hz, H-2α), 1.35 (bs, 3H, Me-14) 1.33 (d, 3H, J = 7.2 Hz, Me-13), 1.16 (d, 3H, J = 7.1 Hz, Me-15). 13C NMR δ 184.5 (C-8), 175.0 (C-12), 167.2 (C-1’), 167.1 (C-10), 150.2 (C-6), 139.3 (C-3’), 136.7 (C-7), 127.4 (C-2’), 124.3 (C-9), 72.4 (C-3), 52.0 (OMe), 43.1 (C-5), 43.0 (C-4), 37.9 (C-11), 32.8 (C-2), 28.1 (C-1), 20.9 (C-5’), 20.2 (C-14), 16.4 (C-13), 15.8 (C-4’) 12.5 (C-15). Data assignment was aided by one- and two-dimensional NMR experiments (Supplemental Material Figure S1).
Computational Methods
The computational methods used are as described previously.19–21
Conformational searching began by generating molecular models of 3S,4R,5R,11R-4 and 3S,4R,5R,11S-4, with their A-rings in both chair and boat conformations, using Spartan’04 software (WaveFunction, Inc., Irvine, CA 92612, USA). The four models were subjected to Monte-Carlo searching using the MMFF94 force field and an energy cutoff of 10 kcal/mol above the global minimum energy. Single-point energy calculations for each conformer were carried out using density functional theory (DFT) at the B3LYP/6-31G* level of theory. The geometries of the conformers found in the initial 5 kcal/mol energy gap were completely optimized using the Gaussian 03 software (Gaussian Inc., Wallingford, CT, USA) at the B3LYP/DGDZVP level of theory. The vibrational modes and rotational strengths of conformers within 2.5 kcal/mol were calculated at the same theoretical level. IR and VCD spectra were calculated by Boltzmann-weighting individual spectra according to the relative free energies of the conformers and considering only those that contribute more than 1% (Table 1). The IR and VCD spectra were calculated using Lorentzian functions and 6 cm−1 bandwidths. The calculated and experimental spectra were compared using CompareVOA software (BioTools Inc., Jupiter, FL, USA) (Table 2).22
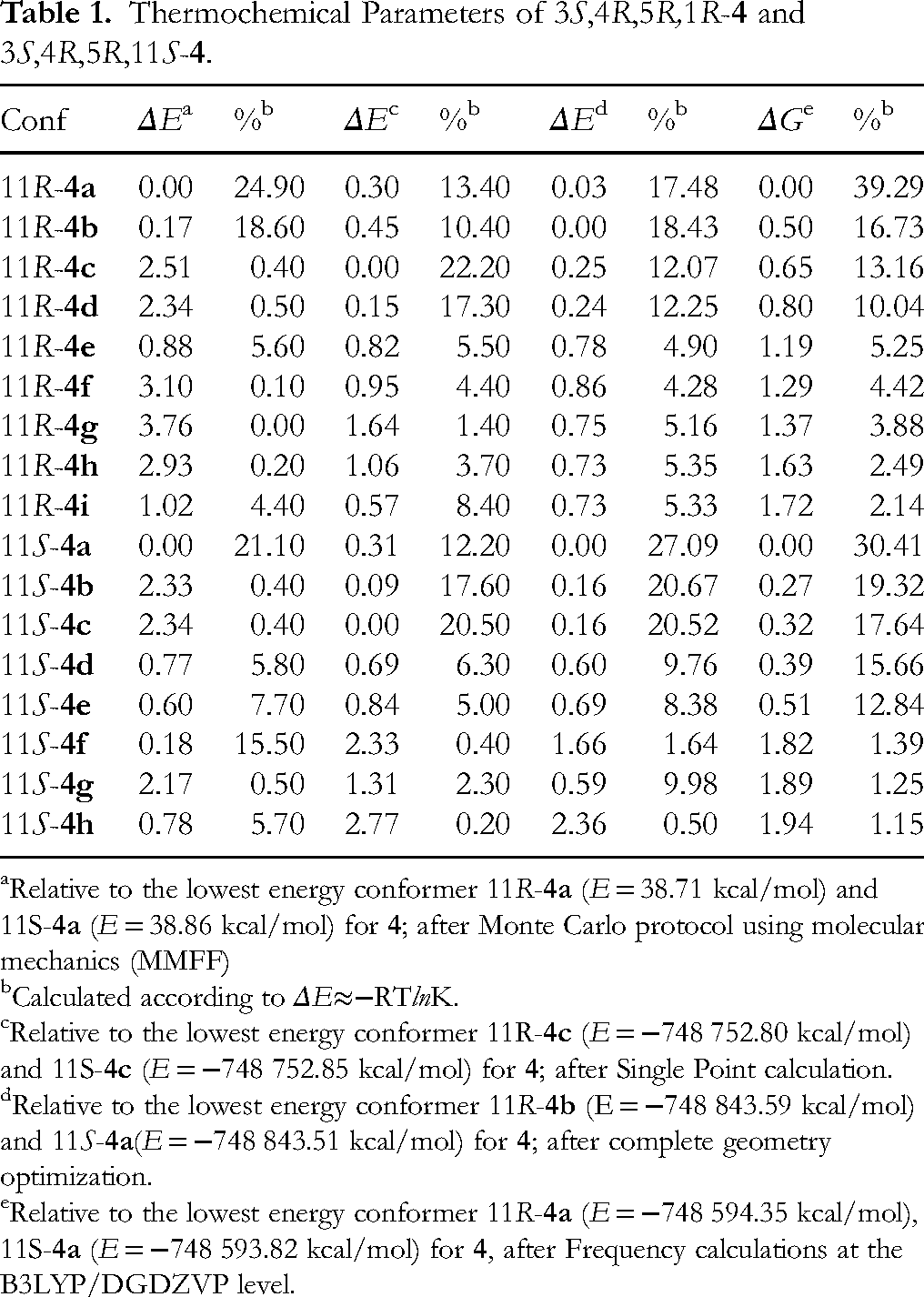
Thermochemical Parameters of 3S,4R,5R,1R-4 and 3S,4R,5R,11S-4.
Conf
ΔEa
%b
ΔEc
%b
ΔEd
%b
ΔGe
%b
11R-4a
0.00
24.90
0.30
13.40
0.03
17.48
0.00
39.29
11R-4b
0.17
18.60
0.45
10.40
0.00
18.43
0.50
16.73
11R-4c
2.51
0.40
0.00
22.20
0.25
12.07
0.65
13.16
11R-4d
2.34
0.50
0.15
17.30
0.24
12.25
0.80
10.04
11R-4e
0.88
5.60
0.82
5.50
0.78
4.90
1.19
5.25
11R-4f
3.10
0.10
0.95
4.40
0.86
4.28
1.29
4.42
11R-4g
3.76
0.00
1.64
1.40
0.75
5.16
1.37
3.88
11R-4h
2.93
0.20
1.06
3.70
0.73
5.35
1.63
2.49
11R-4i
1.02
4.40
0.57
8.40
0.73
5.33
1.72
2.14
11S-4a
0.00
21.10
0.31
12.20
0.00
27.09
0.00
30.41
11S-4b
2.33
0.40
0.09
17.60
0.16
20.67
0.27
19.32
11S-4c
2.34
0.40
0.00
20.50
0.16
20.52
0.32
17.64
11S-4d
0.77
5.80
0.69
6.30
0.60
9.76
0.39
15.66
11S-4e
0.60
7.70
0.84
5.00
0.69
8.38
0.51
12.84
11S-4f
0.18
15.50
2.33
0.40
1.66
1.64
1.82
1.39
11S-4g
2.17
0.50
1.31
2.30
0.59
9.98
1.89
1.25
11S-4h
0.78
5.70
2.77
0.20
2.36
0.50
1.94
1.15
Relative to the lowest energy conformer 11R-4a (E = 38.71 kcal/mol) and 11S-4a (E = 38.86 kcal/mol) for 4; after Monte Carlo protocol using molecular mechanics (MMFF)
Calculated according to ΔE≈−RTlnK.
Relative to the lowest energy conformer 11R-4c (E = −748 752.80 kcal/mol) and 11S-4c (E = −748 752.85 kcal/mol) for 4; after Single Point calculation.
Relative to the lowest energy conformer 11R-4b (E = −748 843.59 kcal/mol) and 11S-4a(E = −748 843.51 kcal/mol) for 4; after complete geometry optimization.
Relative to the lowest energy conformer 11R-4a (E = −748 594.35 kcal/mol), 11S-4a (E = −748 593.82 kcal/mol) for 4, after Frequency calculations at the B3LYP/DGDZVP level.
Cross Comparison of Experimental VCD and IR Features of 4 with the Calculated Spectra for 3S,4R,5R,11R-4 and 3S,4R,5R,11S-4.
anHa
SIRb
SEc
S−Ed
ESIe
Cf
(+)-4versus
3S,4R,5R,11R
0.977
85.8
73.4
13.2
60.2
99
3S,4R,5R,11S
0.977
86.3
63.4
21.3
42.1
82
Anharmonicity factor
IR spectral similarity (%)
VCD spectral similarity for the correct enantiomer (%)
VCD spectral similarity for the incorrect enantiomer (%)
Enantiomer similarity index calculated (SE − S−E)
Confidence level for the AC determination
Statistical analysis was not required for this work.
Results
Furanoeremophilanoid 3 was subjected to oxygenation using continuously flowing oxygen generated in situ from a KI solution and H2O2, while irradiated with a fluorescent lamp and in the presence of methylene blue as a sensitizer. The addition of oxygen to the furane ring11,12,23,24 afforded the butenolactone ring in 2, which was confirmed by comparing its 1H and 13C NMR chemical shifts with those previously reported (Table S2).17 The stereochemistry at C-8 was determined using 1D NOESY experiments, in which selectively irradiating H-6 (δ 4.71) was observed to enhance the 1H NMR signals at δ 5.15, 4.41, and 2.40, which correspond to H-3α, H-8α and H-4α, respectively, thereby confirming that the stereochemistry at C-8 is the same as that described for the natural product.17
Carboxylic acid 1 was obtained by subjecting a solution of 2 in MeOH to alkaline conditions. The mixture was stirred while being irradiated with 60-W microwaves for 90 s at 60 °C to afford 1 and 5 in a 6:4 ratio, as deduced by integrating the 1H NMR signals at δ 6.13 and 6.91 (Figure S1) that correspond to the vinyl protons of the angelate and tiglate group, respectively. The crude reaction mixture was treated with aqueous HCl, extracted with EtOAc, and separated by column chromatography. The 1H NMR spectrum of the product showed signals typical of an angelate ester at δ 6.13 (qq, J = 7.12, 1.5 Hz, H-3’) 2.05 (dq, J = 7.2, 1.5 Hz, Me-4’) and 1.97 (quint, J = 1.5 Hz, Me-5’), and methyl groups at δ 1.35 (s, Me-14), 1.18 (d, J = 7.0 Hz, Me-15), and 1.38 (d, J = 7.2 Hz, Me-13). Signals the correspond to vinylic and methylene protons were observed at δ 6.88 (bs, H-6) and 6.22 (d, J = 1.2 Hz, H-9) and δ 2.71 (dddd, J = 14.3, 13.3, 4.6, 1.3 Hz, H-1β) and 2.35 (ddd, J = 13.1, 3.5, 2.4 Hz, H-1α), 2.24 (dddd, J = 14.4, 4.8, 2.4, 2.4 Hz, H-2β) and 1.68 (dddd, J = 14.4, 14.4, 4.4, 3.2 Hz, H-2α), respectively. The relative configurations of these methylene hydrogens were established using 2D NOESY experiments, in which correlations between Me-14 (δ 1.35) and H-1β (δ 2.71), and H-4α (δ 1.78) with H-2α (δ 1.68) were observed. Methine protons H-3 and H-11 were observed at δ 5.15 (m) and 3.75 (q, 7.2 Hz), respectively. 13C NMR spectroscopy revealed characteristic signals for a cyclic dienone at δ 185.8 (C-8), a carboxylic group at δ 177.3 (C-12), and a carbonyl ester at δ 167.1 (C-1’). HMBC and HSQC experiments were used to assign all NMR chemical shifts. The NMR data are essentially the same as those reported previously (Table S1).1 Although compound 1 was previously described as an oil,1 we attempted to obtain crystals using various solvent mixtures, albeit unsuccessfully. No additional effort was directed toward purifying 5.
The absolute configuration at C-11 was determined by first converting 1 into the corresponding methyl ester 4, which avoids complications related to intermolecular hydrogen bonding.141H and 13C NMR data for 4 (Figure S2) are very similar to those of 1, with the exception that signals at δ 3.76 and 52.0 are present in the 1H and 13C NMR spectra of 4, respectively, consistent with the formation of the methyl ester. In a similar manner to 1, we were unable to crystalize 4.
The protocol for assigning the absolute configuration began by generating molecular models of 3S,4R,5R,11R-4 and 3S,4R,5R,11S-4 and subjecting them to Monte Carlo conformational searching using the MMFF94 force field, as implemented in Spartan’04. Two models were considered for each epimer: the first with the A ring in a chair conformation and the second in a boat geometry. We found 18 and 20 conformers from the chair and boat conformations, respectively, for 11R-4; and 20 and 18 conformers from the 11S-4 epimer. All conformers were subjected to single point energy calculations at the B3LYP/6-31G* level of theory, which revealed that the most stable conformer in each case contains the chair A-ring conformation. The energy difference (ΔE) between the least-stable chair conformation and the most-stable boat conformation of 11R-4 or 11S-4 was calculated to be 4.24 or 4.02 kcal/mol, respectively. Accordingly, only the most stable conformers were considered from this point onwards. The geometries of the selected conformers were fully optimized at the B3LYP/DGDZVP level of theory, which led to 15 and 11 conformers within a 2.5 kcal/mol energy window for epimeric 11R-4 and 11S-4, respectively; these conformers were used to calculate the IR and VCD spectra at the same level of theory. The four most stable conformers of each epimer and their thermochemical data are shown in Figure 2 and listed in Table 1. The final IR and VCD spectra were generated by Boltzmann averaging the contribution of each conformer by considering its Gibbs free energy. Nine and eight conformers of 11R-4 and 11S-4, respectively, were found to contribute to more than 1% of the conformational population (Table 1) and used to generate the averaged spectra (Figures S4 and S5).
The Most Stable B3LYP/DGDZVP Conformers of 3S,4R,5R,11R- and 3S,4R,5R,11S-4.
A visual comparison revealed that the calculated spectra of the 11R isomer better matched the experimental data. Furthermore, we used CompareVOA software22 to compare the experimental spectra of 4 with those calculated for 11R-4 and 11S-4, which revealed a confidence level of 99% for the assigned 11R absolute configuration compared to 88% for the 11S epimer (Table 2).
Discussion
We previously reported isolating furoeremophilanoid 3 and butenolactone 2 for the first time from the roots of Senecio sinuatus.17 To our knowledge, the absolute configuration at C-11 in most natural eremophilanoid carboxylic acids, such as 1, remains undescribed, and has only been determined for a pair of C-11 epimers by X-ray crystallography through the use of bromophenacyl derivatives.15,16 In the reported case, a small 1H NMR shift to higher frequency was observed for the H-11 proton in the 11R epimer. With the aim of expanding our knowledge of the structure of eremophilane sesquiterpenoids, we decided to explore the assigned absolute configuration at C-11 of reported carboxylic acid 11–3 using the VCD approach. For this purpose, 1 was prepared by the oxidation of 3 to afford 2 whose CH-8 configuration was confirmed using NOE 1H NMR spectroscopy. Alkaline hydrolysis of 2 led to 1 along with its tiglate isomerization product 5, which was identified by its typical chemical shifts observed in the 1H NMR spectrum of the crude reaction mixture (Figure S1) at δ 6.91 (qq, J = 7.0, 1.2 Hz, H-3’), 1.84 (dq J = 7.1, 1.1 Hz, H-4’) and 1.90 (quint, J = 1.2 Hz, H-5’). The tiglate isomer was not investigated any further following chromatographic separation. Assuming that the configurations of C-3, C-4, and C-5 of 2 and 3 were not modified throughout the reaction sequence used to produce 1, NOESY experiments enabled us to confirm the Z-configuration of the side chain, the stereochemical relationship between H-3, Me-4, and Me-5, and the alpha and beta configurations of the hydrogen atoms in the CH2-1 and CH2-2 groups (shown in Table S2).
Isomerization of the double bond in the side chain in 2 may lead to 5 during the course of the reaction. Angelate and tiglate groups are widely known to be present in natural products, and some years ago, the controversial assignments of the methyl groups in these esters were settled through 2D 1H/13C NMR correlations observed for their respective acids.25
A speculative reaction mechanism for the formation of 1 is shown in Figure S3. Here, ring opening of the lactone leads to the corresponding carboxylic acid, which, after a series of tautomeric equilibria and the final elimination of a water molecule, yields 1. No product derived from the hydrolysis of the angeloyl group was observed, as evidenced by the lack of signals at around δ 4.1, as observed by us for 3β,6β-dihydroxyeremophyl-1(10)-ene.26 The 11S epimer was not observed following chromatographic separation.
Although the reaction conditions used to generate 4 can possibly epimerize C-11, no evidence of an additional compound was observed, as would have been evident by the presence of an additional H-11 NMR signal with a chemical shift similar to that of the other isomers. The H-11 protons in 1 and 4 exhibited chemical shifts of 3.75 and 3.76 ppm, respectively.
VCD spectroscopy is one of the best independent techniques for determining the absolute configurations of natural products.19–21 After calculating the IR and VCD spectra of the selected conformers and generating population-weighted spectra, 4 was assigned the 3S,4R,5R,11R absolute configuration to a high degree of confidence through comparisons with the experimental data.
As expected, the calculated VCD spectra of 3S,4R,5R,11R-4 and 3S,4R,5R,11S-4 are similar because they are epimers. Although the spectra, at first glance, appeared to correspond better with the experimental data for 3S,4R,5R,11R-4 (Figures S4 and S5), careful analysis of each band in each VCD spectrum using GaussView 5.0 enabled vibrational modes that involve C-11 to be identified (Figure 3). The bands identified as 1-16 correspond to the asymmetric stretching of the bonds in the A and B rings (bands 1, 3, 4, 5, 6, 11, 12, 13, and 15), asymmetric stretching of the bonds in the angelate ester fragment (bands 2, 6, 9, 14), symmetric stretching of the bonds in ring A (bands 9 and 10), asymmetric and symmetric stretching of the carboxylic acid chain (bands 2-8, 11-13 and 15), and the bending motions of the methyl and methine groups (bands 1, 2, 5, 6, 7, 8, 11-16). The vibrational modes involve the entire molecule in almost all cases; consequently, demonstrating differences is difficult. Examples of these vibrational modes are shown in the Supplemental Material (Figures S6 and S7). However, bands 7, 8, and 12, whose signs depend on the C-11 absolute configuration, enable these two epimers to be differentiated. Band 7 at 1164 cm−1 arises from strong CH-11 bending and strong asymmetric stretching motions of the C-11−C-12−O bonds, whereas band 8 at 1184.5 cm−1 corresponds to strong CH-11 bending and strong asymmetric C-12−O−CH3 stretching, along with symmetric bending involving the hydrogen atoms of the methoxy group. Bands 7 and 8 are both positive for the 11R epimer and negative for the 11S epimer. In addition, intense band 12 at 1290.4 cm−1, which is associated with strong CH-11 bending along with symmetric stretching of the C-7−C-11−C-12 bonds, as well as the symmetric bending associated with the hydrogen atoms in the CH3-13 group, is negative for the 11R and positive for the 11S epimer (Figures S6 and S7). CompareVOA software22 was used to determine the degree of similarity between each calculated spectrum and the experimental spectrum of each epimer once these important vibrational modes had been identified. The software uses a numerical method that was developed to facilitate comparisons between acquired and calculated VCD spectra, thereby avoiding personal bias during interpretation. This software also provides an optimal scaling factor that maximizes the similarity between the experimental spectra and the harmonics calculated using hybrid density functionals.27
Comparison between Experimental and Calculated (B3LYP/DGDZVP) VCD Spectra of 3S,4R,5R,11R-4 (Top) and 3S,4R,5R,11S-4 (Bottom).
However, there are some limitations. Although the oxidation reaction using methylene blue as a photosensitizer, the microwave-assisted hydrolysis of 2, and the formation of the methyl ester occurred in short reaction times, the reaction yields were low.
Conclusions
The oxidation of 3β-angeloyloxy-6β-hydroxyfuranoeremophil-1(10)-ene (3) and the subsequent alkaline hydrolysis of the resulting lactone 2 gave 3β-angeloyloxy-8-oxo-eremophila-6,9-dien-12-oic acid (1) with the same stereochemistry as that described for the natural product, as inferred by comparing its 1H NMR spectrum with that previously reported. The absolute configuration of 1, which had been previously isolated from Cacalia species, was determined to be 3S,4R,5R,11R as evidenced by comparing the calculated and experimental vibrational circular dichroism spectra of (+)-4, its methyl ester derivative.
Supplemental Material
sj-docx-2-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-2-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Supplemental Material
sj-docx-3-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-3-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Supplemental Material
sj-docx-4-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-4-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Supplemental Material
sj-docx-5-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-5-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Supplemental Material
sj-docx-6-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-6-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Supplemental Material
Supplemental Material
Supplemental Material
sj-docx-7-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-7-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Supplemental Material
sj-docx-8-npx-10.1177_1934578X251353303 - Supplemental material for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration
Supplemental material, sj-docx-8-npx-10.1177_1934578X251353303 for Preparing an Eremophilanoid from Cacalia Species and Assigning its Absolute Configuration by Brenda Lucía González-Morales, Miguel Ángel Fuentes-Figueroa and Eleuterio Burgueño-Tapia in Natural Product Communications
Footnotes
Acknowledgments
This work was supported by CONAHCYT (Grant A1-S-17910) and SIP-IPN (Grants 20232017 and 20241896).
ORCID iDs
Miguel Ángel Fuentes-Figueroa
Eleuterio Burgueño-Tapia
Ethical Approval
Ethical approval is not applicable for this article.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Funding
The authors disclose the receipt of the following financial support for this research: This work was supported by Consejo Nacional de Ciencia Humanidades y Tecnología (Grant A1-S-17910) and SIP-IPN (Grants 20232017 and 20241896).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of Informed of Human and Animal Rights
This work does not contain any study involving human or animal subjects.
Trial Registration
Not applicable, because this paper does not contain any clinical trials.
Supplemental Material
Supplemental material for this article is available online.
MaoMYangZJiaZ. New eremophilane sesquiterpenes from Cacalia ainsliaeflora. Planta Med. 2003;69(8):745-749. Doi: https://doi.org/10.1055/s-2003-42793
4.
PinderAR. The chemistry of the eremophilane and related sesquiterpenes. In: Progress in the chemistry of organic natural compounds. Vol. 34 Vienna, Springer;1977:81-186. Doi: https://doi.org/10.1007/978-3-7091-8476-9_2
NovotnýLHeroutVŠormF. On terpenes. CLXVIII. Constitution of petasalbine and hydroxyeremophilenolide, components of Petasites albus L. Rhizomes. Collect Czech Chem Commun. 1964;29(9):2189-2193. Doi: https://doi.org/10.1135/cccc19642189
7.
UlubelenAÖksüzS. Eremophilenolides as oxidative artifacts of furanoeremophilanes. J Nat Prod. 1984;47(1):177-178. Doi: https://doi.org/10.1021/np50031a028
8.
HikinoHHikinoYYosiokaI. Structure and autooxidation of atractylon. Chem Pharm Bull. 1962;10(7):641-642. Doi: https://doi.org/10.1248/cpb.10.641
9.
HoriiZYonedaETanakaTIwataC. Photochemical oxidation. II. Biogenetic-type conversion of ligularol and furanoeremophilane into 6(-hydroxyeremophilenolide and eremophilenolide. Chem Pharm Bull. 1977;25(10):2782-2784. Doi: https://doi.org/10.1248/cpb.25.2782
10.
NayaKKanazawaRSawadaM. The photosensitized oxygenation of furanoeremophilanes. I. The isomeric hydroperoxides from petasalbin and their transformations to lactones. Bull Chem Soc Jpn. 1975;48(11):3220-3225. Doi: https://doi.org/10.1246/bcsj.48.3220
11.
LiYSWangZTLuoSDLiSSZhuDY. Sensitized photooxidation of furanoeremophilane with singlet oxygen and their biogenetic relationship. Nat Prod Res. 2006;20(8):724-730. Doi: https://doi.org/10.1080/14786410500185733
12.
NayaKShimizuMNishioHTakedaMOkaSHirotaK. The photosensitized oxygenation of furanoeremophilanes. III. The transformations of the skeletally isomeric lactones from furanofukinol. Bull Chem Soc Jpn. 1991;64(4):1071-1080. Doi: https://doi.org/10.1246/bcsj.64.1071
13.
ToriM. Cumulative data of 1H and 13C NMR signals and specific rotations of eremophilane sesquiterpenoids. 1. Bicyclic eremophilanes (1). Nat Prod Commun. 2022;17(8):1-67. Doi: https://doi.org/10.1177/1934578X221109527
14.
Burgueño-TapiaEJoseph-NathanP. Absolute configuration of eremophilanoids by vibrational circular dichroism. Phytochemistry. 2008;69(11):2251-2256. Doi: https://doi.org/10.1016/j.phytochem.2008.05.011
15.
ToriMWatanabeAMatsuoS, et al.Diversity of Ligularia kanaitzensis in sesquiterpenoid composition and neutral DNA sequences. Tetrahedron. 2008;64(19):4486-4495. Doi: https://doi.org/10.1016/j.tet.2008.02.036
16.
SaitoYIchiharaMOkamotoYGongXKurodaCToriM. Twelve new compounds from Ligularia melanothyrsa; isolation of melanothyrsins A-E, normelanothyrsin A, and other eremophilane sesquiterpenoids. Tetrahedron. 2014;70(16):2621-2628. Doi: https://doi.org/10.1016/j.tet.2014.02.080
17.
Burgueño-TapiaELópez-EscobedoSGonzález-LedesmaMJoseph-NathanP. A new eremophilanolide from Senecio sinuatus gilib. Magn Reson Chem. 2007;45(6):457-462. Doi: https://doi.org/10.1002/mrc.1990
18.
NeisesBSteglichW. Simple method for the esterification of carboxylic acids. Angew Chem Int Ed Engl. 1978;17(7):522-524. Doi: https://doi.org/10.1002/anie.197805221
19.
del RíoREJoseph-NathanP. Vibrational circular dichroism absolute configuration of natural products from 2015 to 2019. Nat Prod Commun. 2021;16(3):1-30. Doi: https://doi.org/10.1177/1934578X21996166
20.
Burgueño-TapiaEJoseph-NathanP. Vibrational circular dichroism: recent advances for the assignment of the absolute configuration of natural products. Nat Prod Commun. 2015;10(10):1785-1795. Doi: https://doi.org/10.1177/1934578X1501001036
21.
Joseph-NathanPGordillo-RománB. Vibrational circular dichroism absolute configuration determination of natural products. In: Progress in the chemistry of organic natural compounds. Vol. 100. Springer International Publishing; 2015: 311-451. Doi: https://doi.org/10.1007/978-3-319-05275-5_4
22.
DebieEGussemEDDukorRKHerreboutWNafieLABultinckP. A confidence level algorithm for the determination of absolute configuration using vibrational circular dichroism or Raman optical activity. ChemPhysChem. 2011;12(8):1542-1549. Doi:https://doi.org/10.1002/cphc.201100050
Joseph-NathanPWesenerJRGüntherH. A two-dimensional NMR study of angelic and tiglic acid. Org Magn Reson. 1984;22(3):190-191. Doi: https://doi.org/10.1002/mrc.1270220312
26.
Velasco-AzorsaRZeferino-DíazRAlvarado-RodríguezJG, et al.Nematicidal activity of furanoeremophilenes against Meloidogyne incognita and Nacobbus aberrans. Pest Manag Sci. 2022;78(6):2571-2580. Doi: https://doi.org/10.1002/ps.6888
27.
AlecuIMZhengJZhaoYTruhlarDG. Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J Chem Theory Comput. 2010;6(9):2872-2887. Doi: https://doi.org/10.1021/ct100326h
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.