
Abstract
Introduction
Triple-negative breast cancer (TNBC) is a global health challenge that accounts for 15–20% of all breast cancers. 1 It is characterised by a lack of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2 (HER2), making TNBCs non-responsive to hormonal or HER2-targeted therapies. 2 Although patients with TNBC respond better to taxanes and anthracyclines than patients with other breast cancer subtypes, the prognosis remains poor owing to the highly metastatic nature of TNBC. Additionally, over 50% of patients who seem to respond to treatment experience relapse in the first 3–5 years after diagnosis. 3
Research has implicated the nuclear factor (NF)-κB signalling pathway in TNBC pathogenesis. Indeed, the NF-κB-target gene SP1 is overexpressed in TNBC and correlates with poor prognosis in patients receiving adjuvant doxorubicin treatment. 4 Moreover, targeting the NF-κB pathway with chemical inhibitors and RNA interference reduced TNBC cell proliferation. 5 NF-κB signalling can be activated by molecules such as interleukin-1 (IL-1), lipopolysaccharides, and tumour necrosis factor-α (TNF-α). 6 In addition, some standard anticancer agents can induce NF-κB signalling, leading to inflammation with resultant organ damage.7,8 For example, cisplatin causes nephrotoxicity, neuropathy, and ototoxicity,9,10 whereas doxorubicin (DOX) causes cardiomyopathy via the NF-κB signalling pathway.11,12 In addition to NF-κB-mediated organ damage caused directly by chemotherapeutics, cancer cells trigger an NF-κB-mediated autocrine feedforward loop that promotes proliferation and metastasis after chemotherapy withdrawal. 13 NF-κB signalling is often activated in TNBC cells, leading to the increased expression of pro-survival and pro-inflammatory genes. This activation contributes to the proliferation, survival, and resistance of tumor cells to apoptosis. 14 NF-κB activation supports tumour growth and invasion through the expression of inflammatory molecules. 15 Furthermore, NF-κB activation in TNBC promotes self-renewal and is associated with chemotherapy resistance. Studies have reported that the activation of the canonical NF-κB pathway in breast cancer cells is associated with doxorubicin resistance.16–18 High NF-κB levels have also been associated with tamoxifen resistance. 16 Given the role of NF-κB in breast cancer progression and chemoresistance, targeting NF-κB has become an area of interest for breast cancer drug development. Given the importance of NF-κB signalling in the progression of TNBC, treatment with NF-κB targeted therapy has been recommended. 5
Amidst these challenges, there is growing interest in natural compounds that can modulate NF-κB signalling and potentially alleviate chemotherapy-induced inflammation. Caffeic acid phenethyl ester (CAPE), an ester of caffeic acid and phenethyl alcohol, is a bioactive component of the New Zealand honeybee propolis. Its pharmacological properties include anticancer, antioxidant, anti-inflammatory, neuroprotective, antimicrobial, and immunomodulatory activities.19,20 CAPE was chosen for this study due to its reported selective cytotoxicity towards specific cancer cells, including breast cancer cells, and its known ability to interfere with NF-κB signalling pathways. The relevance of CAPE to TNBC is underscored by its potential to suppress inflammation and inhibit cancer cell proliferation, migration, and metastasis, all critical factors in TNBC progression and resistance to treatment.
Previous studies have shown the influence of CAPE on cell viability, inducing growth arrest and apoptosis in various cancer cell lines such as breast cancer, 21 colon cancer, 22 melanoma, 23 and glioma cells. 24 The anticancer effects of CAPE have been attributed to its ability to interfere with NF-κB signalling.25–27 CAPE has also been shown to be a potential regulator of NF-kB activity in both in-vivo and in-vitro models of diseases such as calcific aortic valve disease, myocardial ischemia, diabetes, and prostate carcinoma. However, there is a lack of information regarding the effect of CAPE on NF-kB and its target genes, particularly pro-inflammatory cytokines, which are involved in the progression and invasiveness of TNBC cells.28–32 Owing to its antioxidant and anti-inflammatory properties, CAPE can alleviate the detrimental effects of inflammation induced by cisplatin by alleviating the changes in the kidney tissue's TNF- α and IL-6 levels. 33 Another study examining the anti-inflammatory effect of CAPE on activated astroglial cells found that CAPE pre-treatment abrogated NF-κB transcriptional activity in astroglial cells by inhibiting TNF-α induced expression of chemokine (C-C motif) ligand 2 (CCL-2) and intercellular adhesion molecule-1 (ICAM-1) in a cell-specific manner. 34
Nonetheless, the effect of CAPE on cytokine- or chemotherapy-induced NF-κB activation and inflammation in TNBC has not yet been investigated. This study aimed to demonstrate that CAPE suppresses TNF-α- and DOX-induced inflammation and metastasis in TNBC by inhibiting NF-κB signalling. In our study, CAPE reduced cell viability in a dose- and time-dependent manner, indicating its potential as an effective anticancer agent. Additionally, CAPE was shown to inhibit the transcriptional activity of NF-κB, thereby reducing the expression of pro-inflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α as well as inhibiting metastatic markers such as MMP9, MMP2, Vimentin, and VCAM. This study demonstrated that CAPE suppresses TNF-α- and DOX-induced inflammation and metastasis in TNBC by inhibiting NF-κB signalling. These findings highlight the potential of CAPE in TNBC adjuvant therapy, providing solid reasons for its selection and relevance to addressing the aggressive nature of TNBC and its resistance in conventional treatments.
Materials and Methods
Cell Lines and Culture
MDA-MB-231 and MDA-MB-468 cells, obtained from the American Type Culture Collection, were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin-glutamine (all purchased from Gibco-life Technologies, Carlsbad, CA, USA), at 37 °C in a humidified atmosphere containing 5% CO2.
Compounds/Drugs
This study used CAPE (B1644-APE: BIOZOL Diagnostica Vertrieb GmbH, Germany), recombinant human TNF-α protein (210-TA-005/CF: R&D Systems, Minneapolis, MN, USA), and DOX (D1515-10MG: Sigma-Aldrich, St. Louis, MO, USA). The compounds were prepared according to the manufacturer's instructions. CAPE and DOX were dissolved in dimethyl sulfoxide, whereas TNF-α was reconstituted in 1× phosphate-buffered saline (PBS).
Cell Viability Assay
The effects of CAPE, DOX, and TNF-α on MDA-MB-231 and MDA-MB-468 cell viability were determined using an MTT assay. Briefly, the cells were seeded into 96-well plates at a density of 1 × 104 cells/well and incubated at 37 °C for 24 h. The cells were treated with final CAPE concentrations of 0, 2.5,5,10,20,40 μM or final DOX concentrations of 0, 0.1, 0.5, 1,5,10 μM for 24, 48, and 72 h in a final volume of 100 μL of DMEM; or TNF-α (0,0.1,1,5,15 μM) for 24 h and 48 h. Controls included a baseline control of cells with media represented as 0μM with no doxorubicin, CAPE, or TNF-α treatment. Then 20 μL of 2.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St Louis, MO, USA), was added to each well and incubated at 37 °C for 4 h. Subsequently, 100 μL of acidified isopropanol was added to each well and incubated at 37 °C for 30 min. The absorbance was read at 570 nm with a Varioskan™ LUX multimode microplate reader (Thermo Fisher Scientific, Carlsbad, CA, USA). The percent cell viabilities and IC50s were calculated from the absorbance values.
Wound Healing Assay
The ability of MDA-MB-231 and MDA-MB-468 cells to close as an in vitro wound was evaluated to determine the effect of the compounds on cell migration. Briefly, 70 μL of cell suspension containing 2 × 104 cells was seeded into each chamber of a 35 mm μ-dish with culture-insert (ibidi GmbH, Munich, Germany), placed in a 6-well plate, and incubated at 37 °C for 24 h. Cells in the TNF-α-/CAPE group were treated with 10 ng/mL of TNF-α for 24 h. The inserts were removed, and MDA-MB-231 and MDA-MB-468 cells were treated with final concentrations of CAPE (15.5 µM and 11.0 µM respectively) or DOX (0.80 µM) or TNF-α (10 ng/mL) for 48 h. Cells with media only without any treatment were used as untreated controls. Cell migration was monitored using an OPTIKA® microscope (Ponteranica, Italy) at 0, 24, 48, and 72 h. The images were analysed using the ImageJ software (NIH, Bethesda, MD, USA) to determine the percentage of closed cell-free gaps.
Prediction of the Gene Targets of CAPE
The putative targets of CAPE were obtained from the Search Tool for Interactions of Chemicals (STITCH) web server 35 (http://stitch.embl.de/, accessed on 13 August 2022). STITCH integrates information about interactions from metabolic pathways, crystal structures, binding experiments, and drug–target relationships. Inferred information from phenotypic effects, text mining, and chemical structure similarity is used to predict chemical relations. A probability score of 0.7 was used as a threshold for the target search.
NF-κB Target Gene Prediction
The UCSC Gene Interactions tool 36 (https://genome.ucsc.edu/cgi-bin/hgGeneGraph, accessed on August 13 2022) was used to predict the genes that interact with the NF-κB transcription factor. The UCSC Gene Interactions tool displays a detailed gene interaction graph based on data collected from two sources: curated pathway/protein-interaction databases and interactions found through text mining of PubMed abstracts. The tool employs an integrative data source from protein interaction databases such as iRefIndex 13, Androgen Responsive Gene Database, String 9.1, and Negatome 2.0, as well as pathway databases such as KEGG, NCI Pathway Interaction, BioCarta, Reactome 2014, and WikiPathways databases. The predicted gene targets were further confirmed using HitPredict 37 (http://www.hitpredict.org/, accessed on August 13 2022), a resource of experimentally determined protein-protein interactions.
Reverse-Transcription Quantitative PCR (RT-qPCR)
The cells were seeded in 6-well plates at a density of 1 × 106 cells/well and treated as described below. MDA-MB-231 and MDA-MB-468 cells were treated with final concentrations of CAPE (15.5 µM and 11.0 µM respectively) or DOX (0.80 µM) or TNF-α (10 ng/mL) for 48 h. Untreated cells were used as controls. Total RNA was extracted using a Quick-RNA™ Mini-Prep Plus Kit (R1057: Zymo Research, Irvine, CA, USA) per the manufacturer's protocol. RT-qPCR was performed using the Luna Universal One-Step RT-qPCR kit (New England Biolabs, Ipswich, MA, USA) to determine the mRNA expression of TNF-α, IL-8, IL-6, IL-1β, VCAM, vimentin, MMP2 and MMP9. The QuantStudio™ 5 RT-PCR system (Thermo Fisher Scientific, Carlsbad, CA, USA) was used for reverse transcription and amplification. Thermocycling conditions were reverse transcription (55 °C for 15 min), initial denaturation (95 °C for 1 min), 40 cycles of denaturation (95 °C for 15 s), annealing (different temperatures depending on the target gene for 15 s), and extension (60 °C for 1 min). The housekeeping gene GAPDH was used as an internal control. The sequences of the primers used are listed in Supplementary Table S1. QuantStudio™ Design & Analysis Software (Life Technologies, Carlsbad, CA, USA) was used to obtain CT values, and the 2−ΔΔCT method 38 was used to determine the relative expression of each gene.
Statistical Analyses
Data were analysed using GraphPad Prism 9.1.2 (GraphPad Software, San Diego, CA, USA). One-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test was used to compare differences between groups. Data are presented as the mean ± standard deviation (SD) of at least three independent experiments performed in triplicate. Differences between the groups were considered statistically significant when p ≤ 0.05.
Results
Effects of CAPE, DOX, and TNF-α on TNBC Cell Proliferation
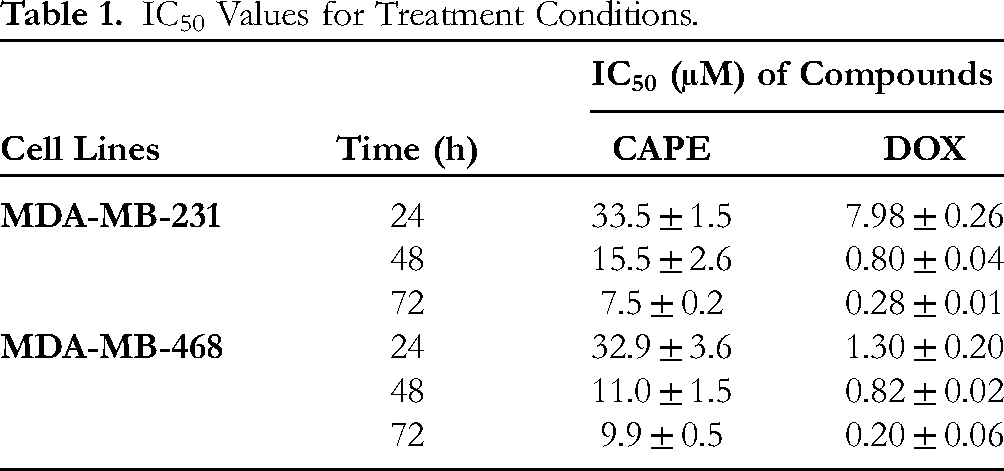
To investigate the influence of CAPE, DOX, and TNF-α on TNBC cell proliferation, an MTT assay was performed. CAPE and DOX reduced cell viability in a dose- and time-dependent manner in both the cell lines (Figure 1A, B). The IC50 values in Table 1 indicate that DOX is more cytotoxic than CAPE. For further experiments, MDA-MB-231 and MDA-MB-468 cells were treated with 15.5 µM and 11.0 µM of CAPE, respectively.
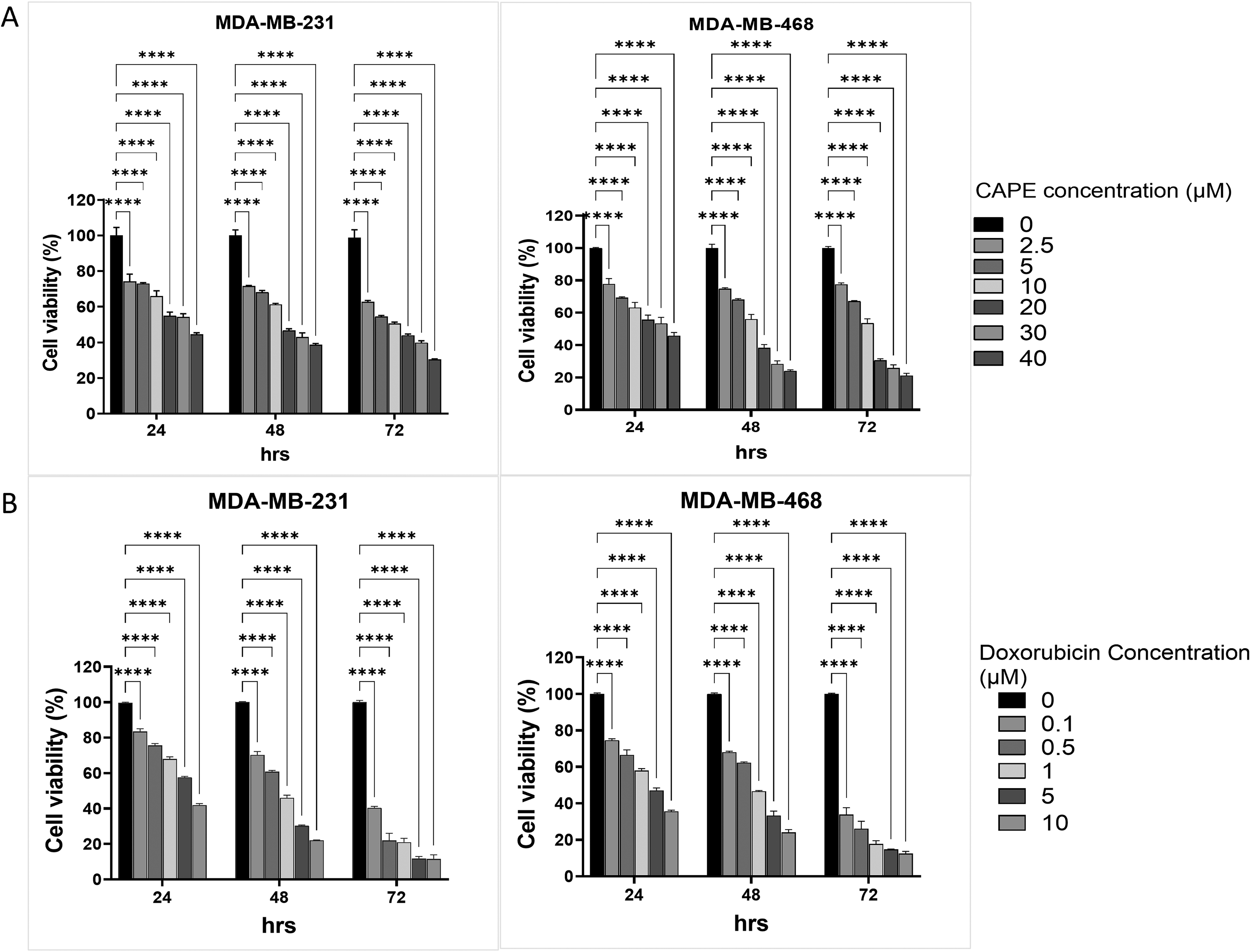
Effects of CAPE, DOX, and TNF-α on TNBC cell proliferation. MDA-MB-231 and MDA-MB-468 cells were treated with increasing concentrations of (
IC50 Values for Treatment Conditions.
CAPE Suppresses TNF-α-Induced TNBC Cell Proliferation
From our studies, TNF-α significantly increased cell viability in both MDA-MB-231 and MDA-MB-468 cell lines in a dose-dependent manner at 24 h and 48 h (Figure 2A), and increasing the concentration of TNF-α increased cell proliferation. Whereas CAPE significantly suppressed TNF-α-induced cell proliferation in both cell lines, with higher concentrations of CAPE leading to an increased reduction in cell viability (Figure 2B). The IC50s of CAPE for TNF-α stimulated MDA-MB-231 cells were 12.0 ± 1.7 µM and 7.7 ± 0.2 µM for 24 h and 48 h, respectively. The IC50s of CAPE for TNF-α stimulated MDA-MB-486 cells were 30.9 ± 1.1 µM and 18.2 ± 1.2 µM for 24 h and 48 h, respectively.

CAPE suppresses TNF-α-induced TNBC cell proliferation. MDA-MB-231 and MDA-MB-468 cells were treated with CAPE for 24 h and 48 h in the presence of TNF-α. Cell viability was determined using an MTT assay. Data are presented as mean ± SD of three independent experiments performed in triplicate. **** p ≤ 0.0001 versus untreated. ns: not significant.
Predictive Analysis of NF-κB Target Genes Modulated by CAPE
Given that CAPE mediates its anticancer activity by interfering with NF-κB signalling,27,39 we sought to identify NF-κB target genes whose expression could be modulated by CAPE. First, we used the STITCH predictive model to demonstrate that CAPE potentially targets multiple proteins, including ReIA and BCL10. ReIA is a transcriptionally active subunit of NF-κB 40 whereas BCL10 is an activator of NF-κB. 41 The activation of these proteins induces NF-κB target gene expression, which regulates cell proliferation, inflammation, and metastasis. 42 Therefore, CAPE may directly inhibit NF-κB activity by interacting with ReIA or indirectly by interacting with BCL10 (Figure 3A). We also used the UCSC Gene Interactions and HitPredict tools to identify NF-κB target genes. The prediction identified IL-1β, IL-6, IL-8, and TNF-α among the top 10 NF-κB target genes (Figure 3B). These genes encode cytokines that promote inflammation in the breast cancer tumour microenvironment.43,44

Predictive analysis of NF-κB target genes modulated by CAPE. (A) A network of molecular interactions is presented. Arrows of different colours represent different edge types: binding (blue), activation (green), inhibition (red), catalysis (magenta), same activity (cyan), and reaction (black). The lines indicate activation, inhibition, or unspecified effects. (B) Predictive analysis of top 10 genes transcribed by ReIA. Arrows show the directionality of interaction based on databases and text mining. Double-headed arrows show that ReIA and target genes interact with each other.
CAPE Inhibits TNF-α and DOX-Induced Activation of NF-κB Signalling
To further confirm the impact of CAPE on NF-κB signalling in TNBC, we examined the effect of CAPE on TNF-α-mediated activation of NF-κB signalling. 45 TNF-α did not significantly affect IL-1β expression in MDA-MB-231 cells but significantly upregulated IL-1β expression in MDA-MB-468 cells (Figure 4A). Additionally, TNF-α induced the expression of IL-6, IL-8, and TNF-α in both TNBC cell lines (Figure 4B-D). CAPE reduced the basal levels of all four cytokines (ie IL-1B, IL-6, IL-8, and TNF-α) as well as their levels in TNF-α-stimulated cells (Figure 4A-D), confirming that CAPE suppresses NF-κB signalling in TNBC.

CAPE inhibits TNF-α and DOX-induced expression of pro-inflammatory cytokines. MDA-MB-231 and MDA-MB-468 cells were treated with CAPE (15.5 µM and 11.0 µM respectively) or DOX (0.80 µM) for 48 h in the absence or presence of TNF-α (10 ng/mL). The mRNA levels of (
Similar to TNF-α, the FDA-approved anticancer agent DOX has been shown to trigger NF-κB signalling. 46 Considering this, we accessed the potential of CAPE to repress DOX-induced NF-κB signalling in the TNBC cells. Except for TNF-α expression in MDA-MB-468 cells, DOX-induced the transcription of IL-1B, IL-6, and IL-8 in both TNBC cell lines, further corroborating its contribution to inflammation in the breast cancer TME. Interestingly, CAPE significantly reduced the DOX-induced expression of all four pro-inflammatory cytokines in both TNBC cell lines (Figure 4A–D). These findings further confirm that CAPE acts by modulating NF-κB signalling.
CAPE Represses Epithelial-Mesenchymal Transition (EMT) in TNBC and TNBC Cell Invasiveness
Tumor metastasis is a multistep process that involves landmark events such as EMT, in which cancer cells become highly motile. 47 Because cytokines such as IL-6, IL-8, and TNF-α can activate pathways involved in EMT, 48 the effect of CAPE on two EMT genes, Vimentin and VCAM1, was examined. CAPE suppressed the basal expression of vimentin in both TNBC cells. Stimulating the cells with TNF-α increased vimentin expression in both TNBC cell lines. Furthermore, DOX did not significantly affect vimentin levels in MDA-MB-231 cells but induced vimentin expression in MDA-MB-468 cells. CAPE also suppressed vimentin expression in TNF-α–stimulated and DOX-treated cells (Figure 5A). Furthermore, TNF-α increased VCAM1 expression in MDA-MB-468 cells but not MDA-MB-231 cells. DOX increased VCAM1 expression in MDA-MB-231 cells but not MDA-MB-468 cells. Treatment of the TNF-α–stimulated and DOX-treated cells with CAPE significantly reduced the expression of VCAM1 (Figure 5B). These observations suggest that CAPE represses EMT in TNBC cells by downregulating vimentin and VCAM. Cancer cells release matrix metalloproteinases (MMPs) during metastasis, which degrade the extracellular matrix to aid their dissemination and invasion. 49 To further unravel the anticancer potential of CAPE, we investigate its influence on the expression of two invasion markers, MMP2 and MMP9. CAPE suppressed basal expression of MMP2 and MMP9 in both cell lines. TNF-α did not affect MMP2, and MMP9 expression in both cells, suggesting that the TNF-α/NF-κB signalling axis may not necessarily contribute to cancer cell invasion. Furthermore, DOX upregulated MMP2 and MMP9 expression in the TNBC cells, and CAPE inhibited DOX-induced expression of these genes (Figure 5C, D).

CAPE represses epithelial-mesenchymal transition (EMT) and cell invasion markers in TNBC. MDA-MB-231 and MDA-MB-468 cells were treated with CAPE (15.5 µM and 11.0 µM, respectively) or DOX (0.80 µM) for 48 h in the absence or presence of TNF-α (10 ng/mL). The mRNA levels of (
CAPE Reduces TNBC Cell Migration
A wound healing assay was performed to measure the cell migratory potential after CAPE treatment. The TNBC cells were treated with the IC25 of CAPE in the presence or absence of TNF-α, and cell migration was monitored for 72 h. Activating NF-κB signalling with TNF-α increased TNBC cell migration, supporting evidence that NF-κB signalling promotes breast cancer cell migration. 50 Furthermore, CAPE inhibited cell migration in unstimulated and TNF-α-stimulated cells (Figure 6A,B).
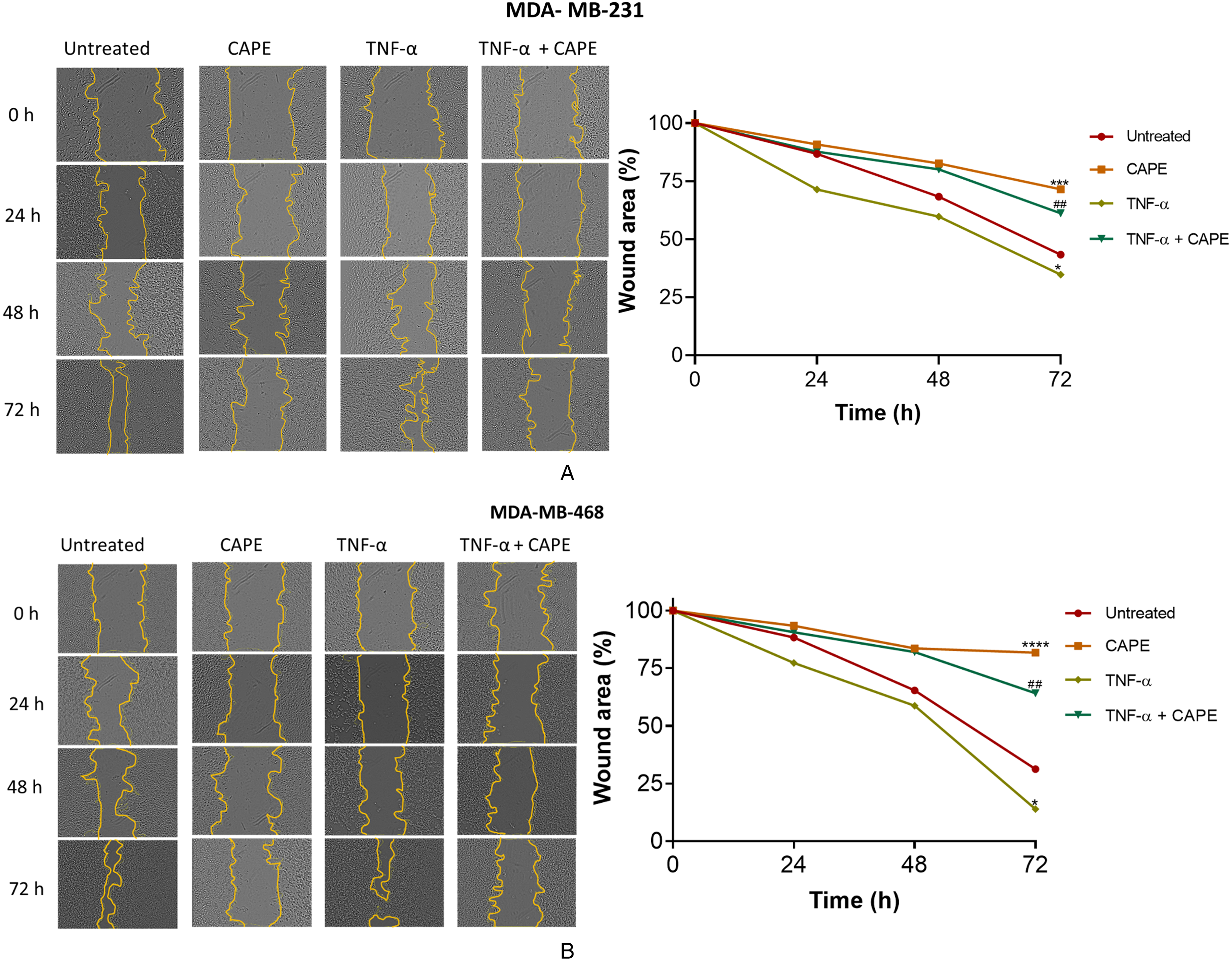
CAPE suppresses the migration of TNBC cells. A. MDA-MB-231 and B. MDA-MB-468 cells were treated with CAPE (15.5 µM and 11.0 µM, respectively) for 72 h in the absence or presence of TNF-α (10 ng/mL). The wound area at each time point is expressed as a percentage of 0 h. Original magnification ×100. Data are presented as mean ± SD of three independent experiments performed in triplicate. ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 versus untreated. # p ≤ 0.05, ## p ≤ 0.01, ### p ≤ 0.001, #### p ≤ 0.0001 versus TNF-α. CAPE: caffeic acid phenethyl ester; ns: not significant.
Discussion
TNBC remains the most difficult-to-treat breast cancer subtype owing to its aggressive nature, lack of classical cell surface receptors, and high propensity for drug resistance. Research has established the contribution of aberrant NF-κB signalling in TNBC pathogenesis, 51 underscoring NF-κB as a critical target for breast cancer therapy. In addition to its canonical activators, the NF-κB pathway can also be activated by cytokines or anticancer drugs.52,53 This study examined the effect of CAPE, an anti-inflammatory compound, on TNF-α or DOX-induced inflammation in TNBC.
In this study, CAPE reduced the viability of both cell lines and was more potent against MDA-MB-468 cells. This observation is consistent with previous studies that reported that CAPE is cytotoxic against breast cancer cell lines.54–56 On the other hand, TNF-α induced proliferation in both TNBC cells, which is consistent with findings that TNF-α promotes breast cancer cell proliferation through positive feedback activation of the TNFR1/NF-κB signalling axis.45,57 Moreover, CAPE suppressed TNF-α-induced TNBC cell proliferation, suggesting that CAPE interferes with the TNFR1/NF-κB signalling axis.
The activation of the TNFR1/NF-κB signalling axis by TNF-α results in a cocktail of cytokine expression, including IL-1β, IL-6, IL-8, and TNF-α. 58 These cytokines are key molecular signatures of multidrug-resistant breast cancers and are associated with angiogenesis, metastasis, and tumour recurrence.59,60 As expected, TNF-α induced the expression of IL-1β, IL-6, IL-8, and TNF-α in the TNBC cells, supporting evidence of the influence of TNF-α on NF-κB-mediated inflammation. 58 Additionally, treatment of cells with DOX resulted in an effect similar to that of TNF-α. Feng et al demonstrated that DOX activates TLR4 signalling in H9c2 cardiomyocytes, leading to the upregulation in MyD88, NF-κB, IL-1 β, IL-6, and TNF-α expression. 61 Similar findings have been reported for MCF-7 breast cancer cells, 62 corroborating our observation.
Additionally, we found that CAPE suppressed the transcription of IL-1β, IL-6, IL-8, and TNF-α in naïve TNBC cells as well as in cells treated with TNF-α and DOX. Since TNF-α and DOX promote inflammation via NF-κB signalling,57,61,63 we attributed the anti-inflammatory property of CAPE to its NF-κB inhibitory activity. Indeed, CAPE has been shown to interfere with NF-κB activity in U-937 histiocytic lymphoma cells, 64 AGS gastric epithelial cells, 65 SKOV-3 ovarian cancer cells, 39 HCT116 colorectal cancer cells, 27 and human CD4+ T cells. 66 Furthermore, Zhang et al (2022) found that CAPE mitigated DOX-induced cardiotoxicity 67 ; which is mediated by inflammation.68,69 Although the exact mechanism of action of CAPE has not yet been defined, its NF-κB inhibitory activity may be due to the prevention of the nuclear translocation of NF-κB or the blocking of the NF-κB-DNA binding, which subsequently halts the transcription of pro-inflammatory cytokines. Our findings suggest that CAPE can attenuate tumour-promoting inflammation and perhaps DOX-induced cardiotoxicity.
Several steps are involved in tumour metastasis, including EMT. In EMT, cancer cells adapt migratory behaviours such as actin polymerisation, cell-cell attachment loss, and cell polarity.70,71 For EMT to be successful, effectors such as E-cadherin, claudins, and occludins must be downregulated, and vimentin, vascular cell adhesion molecule 1 (VCAM1), fibronectin, and N-cadherin must be upregulated. 72 Coincidentally, pro-inflammatory molecules such as IL-6 and IL-8 promote EMT via NF-κB-mediated upregulation of cell migration markers such as vimentin and cell adhesion molecules such as VCAM1. The overexpression of vimentin and VCAM1 in cancer correlates with accelerated tumour growth, invasion, and poor prognosis.42,73 In this study, TNF-α triggered vimentin expression in both TNBC cells, whereas VCAM1 expression was upregulated in MDA-MB-468 but not MDA-MB-231 cells. This observation supports the finding that vimentin and VCAM1 are expressed in response to TNF-α. 6 DOX also induced vimentin and VCAM 1 expression in MDA-MB-468 and MDA-MB-231 cells. This is consistent with reports that DOX can induce EMT in breast cancer. 46 Notably, CAPE suppressed the basal expression of vimentin and VCAM1 in TNF–α-stimulated and DOX-treated cells, indicating that CAPE inhibits EMT in TNBC cells via the downregulation of vimentin and VCAM1.
Cell migration is a key factor for metastasis. 74 In this study, activating NF-κB signalling with TNF-α increased TNBC cell migration, substantiating the evidence that NF-κB signalling promotes breast cancer cell migration. 42 Furthermore, CAPE remarkably impeded cell migration under both unstimulated and TNF-α-stimulated conditions. Therefore, these findings indicate that CAPE is an effective inhibitor of cell migration, supporting similar observations made in MCF-7 breast cancer cells 55 and SKOV-3 ovarian cancer cells. 39 Furthermore, the effect of CAPE on the expression of two cell invasion markers, MMP2 and MMP9, was investigated. MMPs are involved in the degradation of the extracellular matrix to aid in the invasion and dissemination of cancer cells. 75 This study found that the TNF-α/NF-κB signalling axis does not contribute to cancer cell invasion via the MMPs studied. This was reflected by the inability of TNF-α to affect MMP2 and MMP9 expression in both cell types. Furthermore, DOX upregulated MMP2 and MMP9 expression in the TNBC cells, indicating that DOX may contribute to TNBC cell invasion. Several studies on cardiomyocytes76–78 confirm our findings. In addition, CAPE inhibited the basal and DOX-induced expression of these invasive markers, demonstrating that CAPE can inhibit TNBC invasion. Activation of TNFR leads to the interaction of the E3 ubiquitin ligases with protein kinases, which subsequently leads to phosphorylation of activation of IKKβ that in turn induces the phosphorylation and degradation of IκBα, allowing NF-κB dimers, ReLA(P65) and P50, to translocate to the nucleus and drive the transcription of the target gene. 79 Although studies have shown that CAPE prevents NF-kB p65 translocation and activation in cancer cells, 80 this study represents the first study to detail the effect of CAPE on NF-kB target pro-inflammatory cytokines and metastatic markers in TNBC. CAPE disrupts the TNFR1/NF-κB signalling axis by inhibiting the ReLA subunit of NF-kB dimers. In TNBC cells, the disruption of the TNFR1/NF-κB signalling axis by CAPE leads to a significant decrease in the expression of pro-inflammatory cytokines and metastatic markers. This reduction in cytokine levels can attenuate inflammation, decrease cell proliferation, and inhibit metastatic potential, thereby contributing to the suppression of tumour progression and enhancing the overall anticancer effects of CAPE.
Caffeic acid phenethyl ester (CAPE) shows promise as a complementary treatment for triple-negative breast cancer (TNBC) due to its inhibition of inflammation and metastasis markers via the NF-κB signaling pathway. Clinically, CAPE could be used alongside chemotherapeutics like doxorubicin (DOX) to mitigate inflammation-related side effects, including DOX-induced cardiotoxicity, potentially enhancing treatment tolerance. CAPE's inhibition of NF-κB, a pathway linked to chemotherapy resistance, suggests it could boost the efficacy of existing treatments and combat drug resistance. Additionally, its suppression of metastatic markers such as MMP2, MMP9, vimentin, and VCAM1 indicates potential in reducing TNBC metastasis, recurrence, and improving long-term prognosis. Future research should validate its effectiveness and safety in humans through in vivo studies, and explore personalized regimens for TNBC subtypes. Investigating CAPE's integration with other therapies, including immunotherapies and natural compounds, as well as its applicability to other inflammation-driven cancers, is warranted. CAPE's anti-inflammatory properties make it a promising therapeutic agent for TNBC and potentially other malignancies.
This study highlights the potential of CAPE in suppressing the inflammation and metastasis markers involved in TNBC, focusing on TNF-α and DOX-induced NF-κB signalling. Our findings indicated that CAPE is a promising complementary medicine to TNBC due to its potential as an anti-inflammation and anti-metastastic agent. CAPE could clinically be administered as a complement medication to drugs such as doxorubicin, cisplatin, paclitaxel, and fluorouracil in reducing chemotherapy-induced inflammation and inflammation-related organ toxicity induced by these drugs. Inhibition of NF-κB targeted genes by CAPE suggests that it may be used in enhancing the efficacy of existing treatments since the NF-κB pathway is associated with proliferation and chemoresistance. CAPE's ability to inhibit metastatic markers such as MMP2, MMP9, Vimentin, and VCAM1 implies that CAPE is a drug that may decrease TNBC metastasis and recurrence. Future studies on the effect of CAPE on metastatic prostate cancer, lung cancer, colorectal cancer, and gastric cancer could thus be helpful. There is, therefore, the need for further research that will focus on the validation of CAPE in vivo effectiveness and toxicity. Also, considering that within the two subtypes of TNBC, there is variability, the effectiveness of CAPE across these two TNBC cells thus means has the potential be use across a wide range of cancer, hence future research into other subtypes of breast cancers will give insight into such range of cancers that can be targeted for treatment using CAPE. With further research, personalised treatment regimens incorporating CAPE could be developed based on specific patient profiles and tumour characteristics.
In conclusion, this study demonstrates that CAPE mitigates TNF-α and DOX-induced activation of NF-κB signalling and suppresses TNBC cell proliferation, release of pro-inflammatory cytokines, and metastatic processes (Figure 7). Although the findings of this study are relevant to breast cancer research, we used only two TNBC cell lines. Thus, our findings might not be generalised for all TNBC subtypes. Also, the protein levels of the examined genes were not assessed. Therefore, it cannot be established with certainty whether the effect of CAPE at the transcript level affect protein abundance. However, the ability of CAPE to inhibit the growth and migration of both cell lines, that is, mesenchymal-like (MDA-MB-231) and basal-like (MDA-MB-468), is a favourable outcome as it indicates that despite the subtypes of TNBC, CAPE may be an effective therapeutic option.

The proposed mechanism through which CAPE inhibits TNBC progression via NF-κB signalling. TNF-α triggers a series of intracellular events that culminate in the upregulation of NF-κB target genes. CAPE downregulates the NF-κB genes in TNBC cells, inhibiting proliferation, releasing pro-inflammatory cytokines, and suppressing metastatic processes. The figure was created in BioRender. Fosu, K. (2024) (www.biorender.com).
Supplemental Material
sj-docx-1-npx-10.1177_1934578X241298830 - Supplemental material for Caffeic Acid Phenethyl Ester Suppresses Cytokine- and Chemotherapy-Induced Inflammation in Triple-Negative Breast Cancer via NF-κB Signalling
Supplemental material, sj-docx-1-npx-10.1177_1934578X241298830 for Caffeic Acid Phenethyl Ester Suppresses Cytokine- and Chemotherapy-Induced Inflammation in Triple-Negative Breast Cancer via NF-κB Signalling by Kwadwo Fosu, Jude Tetteh Quarshie, Nicholas Awuku Offei, Alberta Serwaa, Bernardine Tuah, Augustine Kojo Sobo, Justum Nii Kotei Amon, Kwabena Amofa Nketiah Sarpong and Anastasia Rosebud Aikins in Natural Product Communications
Footnotes
Acknowledgements
The authors would like to thank Emmanuel Nii Lamptey, Nelson Edu, and Evans Armaah-Vedjesu of the Department of Biochemistry Cell and Molecular Biology at the University of Ghana for their technical support in performing the RT-qPCR and wound healing experiments.
Author Contributions
Conceptualization, K.F., K.A.N.S and A.R.A.; methodology, K.F., J.T.Q., N.A.O., A.S and A.K.S.; writing—original draft preparation, K.F. and J.T.Q.; writing—review and editing, B.T., K.A.N.S and A.R.A.; supervision, K.A.N.S and A.R.A. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The data presented in this study are available in the article and Supplemental material.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical Approval does not apply to this article.
Funding
Kwadwo Fosu was supported by a WACCBIP-World Bank ACE Masters/PhD fellowship (WACCBIP + NCDs: Awandare). This work was supported by funds from a World Bank African Centres of Excellence grant (WACCBIP + NCDs: Awandare) and a DELTAS Africa grant (DEL-15-007: Awandare). This research was funded in whole, by the Wellcome Trust [DEL-15-007] and the UK Foreign, Commonwealth & Development Office, with support from the Developing Excellence in Leadership, Training and Science in Africa (DELTAS Africa) programme.
WACCBIP+NCDs: Awandare, (grant number DEL-15-007: Awandare).
Statement of Human and Animal Rights
This article does not contain studies with human or animal subjects.
Statement of Informed Consent
No human subjects are in this article, and informed consent is not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.