Objective: The present contribution describes a novel approach for obtaining steroid derivatives of the iso-type from sapogenins for drug discovery. Methodology: Diastereomeric 23-acetyl-spirostanols were prepared by acetolysis of smilagenin (4) (25R) using boron trifluoride diethyl etherate in acetic anhydride followed by treatment of the epimeric epoxycholestene obtained (6 or 9) under basic hydrolysis. Higher regioselectivity was attained in the presence of ZnCl2 in acetic anhydride. Results: Acetolysis of smilagenin (4) (25R) using boron trifluoride diethyl etherate in acetic anhydride afforded 20-α-methyl epoxycholestene 6 as the major product, two furostene derivatives (7 and 8), as well as the new 20-β-methyl epoxycholestene 9. Increased regioselectivity for the acetolysis of smilagenin (4) was observed using ZnCl2 in acetic anhydride affording 6 in 90% yield. Subsequent treatment of the epimeric epoxycholestene (6 or 9) under basic hydrolysis afforded two new diastereomeric 23-acetyl-spirostanols 14 and 15. The structures of all compounds were established using 1D and 2D NMR techniques. Conclusions: Formation of the two diastereomeric 23-acetyl-spirostanols (14 and 15) from epimeric epoxycholestenes 6 and 9 with KOH/EtOH proceeds with high stereospecificity. The configuration at C-20, C-22, C-23, and C-25 for the new iso-type 23-acetyl-spirostanol 15 was established by X-ray crystal analysis as 20R, 22R, 23S, and 25R. In addition to the above, the X-ray of 15 was analyzed and compared with the crystallographic data of sarsasapogenin acetate (16), smilagenin (4) and 23-acetyl- sarsasapogenin (17).
Steroid saponins are glycosides with a wide range of biological activities which are widely distributed in plants.1 Hydrolysis of saponins provides steroidal aglycones known as sapogenins which can be of the furostane, cholestane, or spirostane type, the latter is characterized by the presence of a spiro moiety which connects rings E and F. Most naturally occurring spirostane sapogenins have the R configuration at the spiro carbon (C-22) and an α-oriented C-21 methyl (20S), but they can also differ in the configuration at C-25 as well as exhibit cis or trans junction at rings A/B or the presence of a double bond at C-5 and C-6 (Figure 1).
Steroidal sapogenins from natural sources.
Sapogenins are important precursors for the synthesis of steroidal bioactive drugs. Diosgenin, the most widely used member of this family, has found a wide range of applications in functional food and in care industries, but especially in the pharmaceutical industry.2 Some important drugs synthesized from diosgenin are prednisone, prednisolone, dexamethasone, betamethasone, methylprednisolone, and hydrocortisone which are prepared by a combination of chemical and biological (fermentation) methods.3
The spiroacetal structural motif is present in many bioactive substances, natural products, and drugs, for this reason, spirocyclic compounds have attracted the interest of researchers in various fields of chemistry. Despite their importance and bioactivity, access to molecules having the spiroacetal structural motif has been hampered by the limited stereocontrolled synthetic approaches available. The most common synthetic procedure for preparing spiroacetals is the acid-catalyzed cyclization of ketodiols, and this has been extensively employed in natural product synthesis.4
The presence of a stereogenic center at the spiroacetal carbon of sapogenins gives rise to normal and iso-type structures. Several groups have reported that isomerization of the normal to the iso structures proceeds via pseudo-type intermediates such as those formed during acid hydrolyzes (Figure 2).5a The most common isomerization is that of sarsapogenin (2) to smilagenin (4).1c,5b
Distinction with respect to the E/F rings in spiroacetals and pseudo structure.
It has been reported that the transformation of sapogenins to furostenols proceeds in acetic anhydride solution at the boiling point rather than at 200 °C. Other procedures involve treatment of sapogenins in acetic anhydride with boron trifluoride diethyl etherate although in these cases the yields of furostenes are low.6
It is important to highlight that due to the wide number of applications and remarkable bioactivity of spirostanic sapogenins, as well as their use in the synthesis of steroidal hormones, a great number of studies promoting side chain rearrangement under acidic conditions have been reported. On the other hand, it is well known that sapogenins of the iso-type or those having 20R configuration are rare in natural products and are usually prepared synthetically or by microbial transformation. For this reason, studies on their synthesis and chemical reactivity are scarce. Nonetheless, due to their chemical structure and pharmacological and stereoselective properties, 20R-saponins have attracted a great deal of attention in recent years.1d
Our group has shown that the regioselectivity to ring opening of the E and F rings of sapogenins of the 25R and 25S series varies depending on the Lewis acids used. For example, the acid catalyzed reaction of diosgenin (1) and hecogenin (3), using boron trifluoride diethyl etherate affords the epoxycholestene derivatives with high stereoselectivity.7 In contrast, E/F cleavage of the spiroacetal in sarsasapogenin (2) yields a mixture of epoxycholestene and furostene derivatives.8a
In this context, we have reported that treatment of spirostanic sapogenins with different Lewis or Brønsted-Lowry acids, or even the same acid, produces different steroidal skeletons.8b–c Moreover, the chemical shift values of some of the carbons of the C-20S and C-20R-epimers show regular differences including C-17, C-21, and C-22.
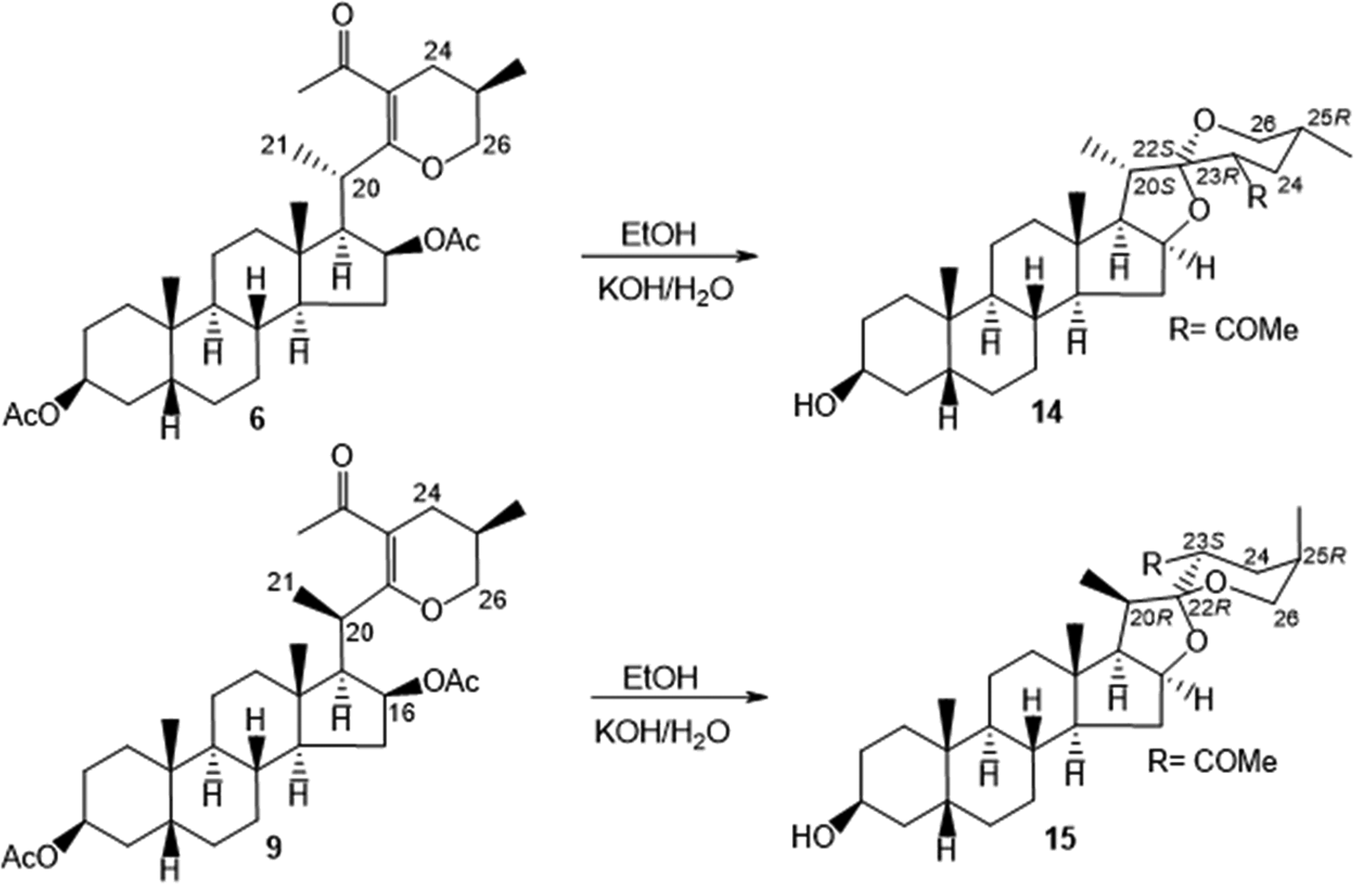
We describe herein the results obtained from the acetolysis of smilagenin (4) using boron trifluoride diethyl etherate and ZnCl2 in acetic anhydride. In agreement with the other acetolysis results from the sapogenins of the 25R series,7,9 cleavage of smilagenin (4) proceeds with high regioselectivity favoring the epoxycholestene (6). Additionally, a new β-methyl oriented epoxycholestene 9 (C-20R) analogous to the product described by Lu for diosgenin (1), sarsasapogenin (2), and tigogenin (5)9 was isolated in small amounts by HPLC. Further transformation of epoxycholestene 6 (C-20S) with KOH/EtOH, under the conditions described for diosgenin (1), sarsasapogenin (2), and tigogenin (5)7c,9 gave the 23-acetyl-spirostanol 14. In turn, the C-20R epimeric epoxycholestenes 9 derivative of smilagenin afforded, after treatment with base, a new iso-type spirostanol 15 with the C-20R, C-22R, C-23S, and C-25R configuration whose structure was established by 1D and 2D NMR experiments and confirmed by X-ray analysis.
Materials and Methods
Experimental Section
1D and 2D 1H and 13C NMR spectra (DEPT, COSY, HMQC, HMBC, INADEQUATE) were recorded on JEOL eclipse +400 MHz, and DMX 500 spectrometers. Chemical shifts are stated in ppm (δ) and referred to the residual 1H signal (δ= 7.27) or to the central 13C triplet signal, (δ= 77.0) for CDCl3. Infrared absorption spectra were obtained using KBr pellets with a Perkin-Elmer 16F-PC-FT-IR spectrophotometer Mass (MS) spectra were obtained on a HP 5989A spectrometer adapted to a HP 6890A.
Ultraviolet absorption spectra were determined on a Perkin-Elmer Lambda 12 spectrophotometer; wavelengths (λ) are expressed in nm. Optical rotations were determined on a Perkin-Elmer 241 polarimeter at room temperature using chloroform solutions. Melting points were determined on an Electrothermal 9200 apparatus. Elemental analyses were performed on a Thermofinnigan flash 1112 and column chromatography was carried out on silica gel grade 60 (230-400 mesh).
Structural data of steroidal sarsasapogenin acetate (16), smilagenin (4), (20R,22R,23S,25R)-23-acetyl-5β-spirostan-3β-ol (15), and 23-acetyl-sarsasapogenin (17)10 were collected on an Enraf Nonius KappaCCD at 293 K with MoKα radiation (λ = 0.7173 Å). The crystals were mounted on conventional MicroLoopsTM. All heavier atoms were found by Fourier map difference and all atoms appear in the first solution, in some compounds the unit cell contains a solvent molecule that does not interfere with the analysis of the molecules under study, modeling solvent disorder makes refinement problematic. All reflection data were corrected for Lorentz and polarization effects. For all crystal structures, the first structure solution was obtained using the SHELXL program and the SHELXL-2019 was applied for refinement and output data.11,12 All software manipulations were performed through the ShelXle program.12 Mercury 2020.1 and ORTEP-3 were used to prepare artwork representations13 CCDC 2250710 (sarsasapogenin acetate 16), 2250712 (smilagenin 4), 2250711 (20R,22R,23S,25R)-23-acetyl-5β-spirostan-3β-ol (15), and 2250713 (23-acetyl-sarsasapogenin 17) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Crystallization Details
Crystals of smilagenin (4), (20R,22R,23S,25R)-23-acetyl-5β-spirostan-3β-ol (15), and 23-acetyl-sarsasapogenin (17) were grown by slow evaporation of a solution of methanol:water (85:15). Sarsasapogenin acetate (16) was crystallized from THF.
Reaction of Smilagenin (4) with ZnCl2/Ac2O
To a solution of 1.00 g (2.40 mmol) of 4 in 10 ml of Ac2O were added 0.30 g (2.4 mmol) of ZnCl2 at room temperature. The mixture was stirred and monitored until complete disappearance of starting material (40 h) and the reaction quenched pouring the reaction over iced water. The organic phase was extracted with ethyl acetate, neutralized with a saturated sodium bicarbonate solution, dried with anhydrous Na2SO4, and evaporated to dryness under vacuum to yield 1.10 g of crude product which was chromatographed over silica gel using hexane/EtOAc (85:15) to give 1.00 g of 6 (90% yield, Rf = 0.61) and hexane/EtOAc (70:30) to give 0.060 g of 7 (4% yield, Rf = 0.28).
Reaction of Smilagenin (4) with BF3·OEt2/Ac2O
To a magnetically stirred solution of 4 (1.00 g, 2.4 mmol) in 10 ml of Ac2O were added 3 ml (10 mmol) of BF3.OEt2, at room temperature. The mixture was stirred and monitored until complete disappearance of the starting material (3 h). The reaction was quenched adding ice and vigorously shaken. The organic phase was extracted with ethyl acetate, neutralized with a saturated sodium bicarbonate solution, dried with anhydrous Na2SO4, and evaporated to dryness under vacuum. The crude product was chromatographed over silica gel using hexane/EtOAc (85:15) to give 0.65 g of a mixture of 6 and 9 (Rf = 0.6, 70% yield in a 9:1 ratio, as determined by HPLC), 20% of (E)-(20S,25R)-20,23-Diacetyl-5β-furost-22-ene-3β,26-diyl diacetate (7) (Rf = 0.10) with hexane/EtOAc (60:40) and traces of 8.
Hydrolysis of the Mixture of Epoxycholestenes 6 and 9
To a 10% ethanolic KOH solution (50 ml) was added the mixture of 6 and 9 (0.50 g, 1.09 mmol) and the solution was vigorously stirred at room temperature for 48 h. After, the solvent was removed under reduced pressure and neutralized with 10% HCl and the organic phase was extracted with EtOAc-H2O, washed several times, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was chromatographed over silica gel using hexane/EtOAc (85:15) to give 0.327g of 14 (78% yield, Rf = 0.47) and with 80:20, 0.100 g of 15 (8% yield, Rf = 0.37).
A fraction enriched with 9 (10 mg, 0.018 mmol) obtained from the mixture of 6 and 9 was hydrolyzed as described above giving 5mg of pure15.
Considering that spirostanic sapogenins exhibit significant reactivity differences depending on the Lewis acid used for cleavage, or even with the same acid, we decided to explore the acetolysis of smilagenin (4) (25R), (Scheme 1). Thus, cleavage of smilagenin under the conditions previously described for diosgenin (1), hecogenin (3) (25R),7a and sarsasapogenin (2) (25S),8a using boron trifluoride diethyl etherate in acetic anhydride afforded (20S,25R)-23-Acetyl-22,26-epoxy-5β-cholest-22-ene-3β,16β-diyl diacetate (6) as the major product (70% yield), and two furostene derivatives: (E)-(20S,25R)-20,23-Diacetyl-5β-furost-22-ene-3β,26-diyl diacetate (7) (20% yield) and (E)-(20S,25R)-23-Acetyl-26-hydroxy-5β-furost-22-ene-3β-yl acetate (8) in trace amounts. HPLC analysis revealed the presence of a new derivative 9 which could not be obtained pure from column chromatography due to the similarities in Rf and the low yield of the product; nonetheless, a small amount of the compound was collected by HPLC and spectroscopically characterized as the β-oriented (20R)-epoxycholestane 9. The analogous derivative from diosgenin was obtained by Tian using BF3·OEt2/Ac2O,14 and recently by Li and Huang9 who reported the preparation of epoxycholestene derivatives with the 20β-methyl configuration from diosgenin (1), sarsasapogenin (2), and tigogenin (5) using Lewis acid catalysis in CH2Cl2 as solvent instead of acetic anhydride.
Products from cleavage of rings E/F of smilagenin (4) using BF3·OEt2.
The selectivity observed for smilagenin (4) is in agreement with the results obtained for sapogenins from the 25R series7a,9 and in marked contrast with the low E/F regioselectivity observed in the cleavage of sarsasapogenin (25S)8a which has been attributed to steric hindrance of the axial methyl group at C-25 over the axial proton at C-23 which hinders β elimination to form the double bond in the dihydropyran that leads to the epoxycholestene (Scheme 1).
The structures of all compounds were established using 1D and 2D NMR experiments. The assignments of the individual chemical shifts were based on the values for sapogenins and similar compounds previously reported.15 Selected 1H NMR chemical shifts for epoxycholestenes derived from diosgenin (1), sarsasapogenin (2), and smilagenin (4) are summarized in Table 1. The β-Methyl epoxycholestene 9 (20R) can be differentiated from the α−methyl 6 epimer by the 1H NMR chemical shift of H-26 (δ 4.06 and 3.35 ppm in 9) which is partially overlapped with H-20 (δ 3.98 ppm) since the same protons in 6 appear at 3.99 and 3.45 ppm. Both compounds show the molecular ion peak at M+ 542 and similar IR bands.
Selected 1H NMR Chemical Shifts for Epoxycholestenes in ppm.
As proposed recently,9 epimerization at C-20 can be explained by formation of a pseudosapogenin intermediate. These results are also in agreement with those of Gould6a who treated diosgenin (1) with a variety of acids at different concentrations and found that low acid concentration promotes the formation of pseudosapogenin.
Increased regioselectivity for the acetolysis of smilagenin (4) was observed using ZnCl2 acetic anhydride as previously described for diosgenin (1)7 yielding the corresponding 20-α-methyl epoxycholestene (6) in 90% yield.
NMR Spectral Analysis
Unequivocal signal assignment in the 1H NMR spectrum for the major product 6 was done with the aid of 2D NMR techniques (Figure S1). The 1H NMR spectrum of 6 shows a td, J = 7.8 Hz, J = 3.0 Hz at 5.11 ppm for H-16 characteristic for a methine containing an acetate group and a broad signal at 5.05 ppm for H-3. The diastereotopic protons at C-26 give rise to the signals at 3.99 (H-26eq) and 3.45 ppm (H-26ax). The signal for H-20 is shifted to high frequencies (4.05 ppm) compared to smilagenin (4) (1.86 ppm), as expected for an enol ether.
The 13C assignment for 6 was done with the aid of COSY, HMQC and HMBC experiments (Figure S2). The DEPT spectrum of 6 allowed to identify 7 methyls, 9 methines, and 10 methylenes with opposite phase (Figure S3). The 13C signals were also assigned by comparison with the chemical shifts of smilagenin (4)11b and reported epoxycholestenes derived from other sapogenins.7a,8a,9,15
The HMQC experiment confirmed the correlation between C-26 at 71.4 ppm with the signals at 3.99 and 3.45 ppm. The methine carbons C-3/C-16 at 70.6 and 75.2 ppm correlated with the multiplet at 5.05 ppm (H-3) and with the td at 5.11 ppm (H-16), respectively. The methyl signals assigned to C-21/C-27 at 1.17 and 0.97 ppm appear as doublets, therefore the singlets at 0.89 and 0.98 correspond to C-18/C-19 (Figure S4).
The HMBC experiment confirmed the correlation between C-22 (171.3 ppm) with CH3-21 (1.17 ppm) and H-20 (4.05 ppm). In turn, the carbonyl at 170.6 ppm (3-OAc) correlates with the proton signal at 2.04 ppm (Figure S5).
Independent confirmation of 13C assignments was obtained by an INADEQUATE experiment which allowed to confirm almost all 13C-13C couplings in epoxycholestene 6 (C-6/C-7 not observed) (Figure 3, only some correlations are marked for clarity).
Aliphatic region of the 13C-13C INADEQUATE for compound 6 in CDCl3 at 125 MHz (only some correlations are marked for clarity).
Selected 1H NMR chemical shifts for compounds 6 and 9 were compared with the 22,26-epoxycholest-22-enes obtained from diosgenin and sarsasapogenin (10a-b and 11a-b).7a,8a,9 Analysis of the data in Table 1 shows that information on the stereochemistry at C-20 in epoxycholestenes can be obtained by comparison of the chemical shifts of CH3-21 and CH3-18 since derivatives with an α methyl at C-20 (20S) give signals between 1.15 and 1.17 ppm for C-21 while CH3-18 appears in the region from 0.87to 0.90 ppm. In turn, for compounds with a β methyl at C-20 (20R), both signals are shifted to low frequency due to steric hindrance and appear between 0.99 and 0.97 ppm for CH3-21 and 0.71- 0.74 ppm for CH3-18. In addition, a significant difference in the chemical shift is observed for 16-OAc (1.80-1.84 ppm for derivatives with α methyl (20S) configuration) compared to those with β methyl (20R) which appear in the range from 1.90 to 2.02 ppm.
Furostene derivatives 7 and 8 were assigned by comparison with the data reported in the literature.7c,8a
Synthesis of 23-acetyl-spirostanols
Having the two C-20 isomeric epoxycholestene derivatives of smilagenin, we decided to explore their reactivity under basic conditions to give stereoisomeric 23-acetyl-spirostanols. Thus, treatment of the mixture as well as each of the epimeric epoxycholestenes 6 and 9 with KOH/EtOH at room temperature7c,10 proceeded with high stereospecificity to give two diastereomeric 23-acetyl-spirostanols (14 and 15) (Scheme 2). The structures of these compounds were determined by NMR, and the configuration at C-20, C-22, C-23, and C-25 for compound 15 was established by X-ray crystal analysis as 20R, 22R, 23S, and 25R. Analogous compounds were obtained by Lu recently,9 however, the stereochemistry at C-22 and C-23 in the analogous 23-acetyl-spirostanols derived from diosgenin, hecogenin, and tigogenin were erroneously assigned as 22S and 23R.
Synthesis of 23-acetyl-spirostanols 14 and 15.
Introduction of the new functionality at C-23 produces the expected downfield shift in both the axial and equatorial protons of H-24. Downfield shifts are also observed for H-20 due to the proximity of the substituent attached to C-26 which causes van der Waals compression of H-20 by the substituent at C-23. Table 2 summarizes the 1H NMR data of selected protons of spirostanols derived from smilagenin (4) obtained in this study, as well as the data reported for analogs from sarsasapogenin (2) and diosgenin (1).9 The high-frequency shift of the C-18 methyl group and C-21 doublet can be attributed to steric compression. The C-18 methyl is shifted towards the methyl resonance of C-19 in agreement with previous reports that the chemical shift of angular methyl groups C-18 and C-19 is influenced by structural variations in the structure.16
Selected 1H NMR Chemical Shifts for Spirostanols in ppm.
The structures of the compounds were analyzed by the comparison of the proton spectra of isomeric 23-spirostanols derived from sarsasapogenin (2 and 12b) and smilagenin (14 and 15). Figure 4 shows that the diastereotopic protons at C-26 resonate in the region from 3.3 to 4.1 ppm (Δδ= 1.2) and show a considerable difference in chemical shifts and splitting patterns depending on the configuration at C-25. Smilagenin (4) and 23-acetyl-smilagenin (14) show a triplet and a doublet of doublet with similar chemical shifts for H-26, typical of equatorial methyls at C-25. However, the same protons show a drastic difference in chemical shifts in sarsasapogenin 2 and 23-acetyl-sarsasapogenin 15 with an axial methyl group at C-2510,17(Figure 4). This allows to establish that 14 and 15 have the methyl group at C-25 oriented in equatorial and axial position, respectively. These results can only be explained by stereospecific nucleophilic attack of the alkoxide at C-16 to C-22 of the epoxycholestene in basic medium. Thus, C-20 alpha epoxycholestenes undergo attack at the Si face while in the C-20β epimer the attack occurs from the Re face.9
1H NMR spectra of smilagenin (4), sarsasapogenin (2) and isomeric spirostanols 12b, and 14-15 in CDCl3.
The 13C chemical shifts for isomeric spirostanols 14 and 15 show no significant differences for compounds with different configuration and were assigned by comparison with literature data and 2D experiments.7c,10,11a,j
Single Crystal-XRD Studies of the Compounds 4 and 15–17
The crystal structures and crystallographic data (Table S1) for sarsasapogenin acetate (16), smilagenin (4), (20R,22R,23S,25R)-23-acetyl-5β-spirostan-3β-ol (15), and 23-acetyl-sarsasapogenin (17) are shown in Figure 5.
ORTEP diagrams for compounds (a) 16 (b) 4 (c) 15 and (d) 17, with thermal ellipsoids drawn with a probability of 50%.
Sarsasapogenin acetate (16), smilagenin (4) and (20R,22R,23S,25R)-23-acetyl-5β-spirostan-3β-ol (15), crystallized in a monoclinic system in the chiral space group P21, with two and four molecules within the unit cell, 23-acetyl-sarsasapogenin (17) crystallized in the orthorhombic system, chiral space group P212121, with 4 molecules within the unit cell. Compounds 16 and 4 crystallized with the presence of solvent, THF, and water, respectively; however, the solvent does not interfere with analysis of the molecules under study.
All compounds studied contain the fused central skeleton of the steroid formed by ABCD rings, presenting cis A/B annular junctions with substituents at C-5 and C-10 oriented β with respect to the annular plane, and cis D/E between the cyclopentane and tetrahydrofuran rings with hydrogens at C-16 and C-17 in α orientation with respect to the annular plane. The B/C and C/D junctions are trans with hydrogens at C-8 and C-13 oriented β with respect to the annular plane and hydrogens at C-9 and C-14 in α with respect to the annular plane.
Sarsasapogenin acetate (16) and smilagenin (4) are structurally similar natural steroid sapogenins. The main structural differences lie in the methyl groups at C-25 of the oxa-spirocyclic fragment, which is axial in sarsasapogenin acetate (16) and equatorial in smilagenin (4) another difference is found in the substituents at C-3, having an acetoxyl group in sarsapogenin and a hydroxyl group in smilagenin, both β with respect to the annular plane.
All six-membered rings (A/B/C/F) for the four compounds have chair conformations without significant disturbances.18 As in similar compounds, the oxygen of the equatorial 3-OAc substituent in sarsasapogenin acetate 16 and 23-acetyl-sarsasapogenin 17 as well as in the 3-hydroxy group in compounds 4 and 15 is arranged in an almost eclipsed disposition to the hydrogen atom which is axial, this arrangement favors that conformational perturbations do not occur in the A ring. The five-membered D rings adopt a twist conformation and ring E adopts an envelope conformation with the oxygen in an exo orientation.
Compound 16 does not exhibit any interaction with another molecule of the same compound in the unit cell (Figure 6a). Similarly, as can be seen in Figure 6b, compound 4 shows no interactions with another molecule in the unit cell, but it shows interaction with the water molecules present, through classic hydrogen bonds in O(04)-H(041)···O(02) (2.784 Å, 161.46°) and O(04)-H(043)···O(3) (2.842 Å, 157.02°).19
Unit cell and short interactions of (a) sarsasapogenin acetate 16 and (b) smilagenin 4.
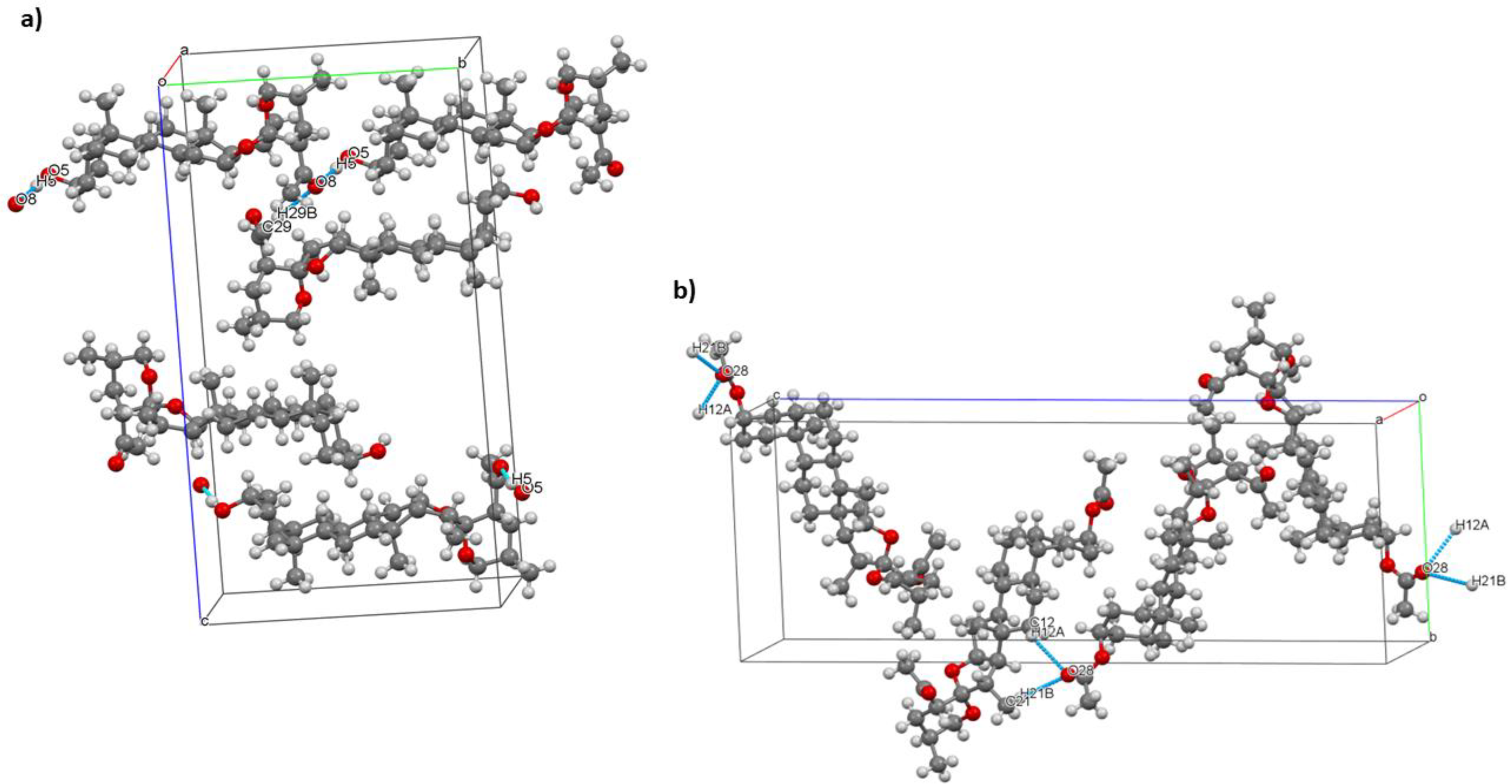
Molecules in the unit cell of compound 15 have a trimeric structure (Figure 7a), these interact through non-classical hydrogen bonds in C(29)-H(29)···O(8) (3.472 Å, 163.03°) and also show interaction via classical hydrogen bonds in O(5)-H(5)···O(8) (2.894 Å, 152.11°). Meanwhile, in the unit cell of compound 17, the molecules form dimers through non-classical hydrogen bonds (Figure 7b) in C(21)-H(21B)···O(28) (3.628 Å, 161.83°) and in C(12)-H(12A)···O(28) (3.340 Å, 125.19°). Non-classical C-H···O hydrogen bonds often display D-H···A distances of up to 3.0 Å and low D-H A angles > 90.19
Unit cell and short interactions of compounds (a) 15 and (b) 17.
Conclusions
The acetolysis of smilagenin (4) (25R) using boron trifluoride diethyl etherate in acetic anhydride, afforded the new (20S,25R)-23-Acetyl-22,26-epoxy-5β-cholest-22-ene-3β,16β-diyl diacetate (6) as the major product (70% yield), and two furostene derivatives, (E)-(20S,25R)-20,23-Diacetyl-5β-furost-22-ene-3β,26-diyl diacetate (7) (20% yield) and (E)-(20S,25R)-23-Acetyl-26-hydroxy-5β-furost-22-ene-3β-yl acetate (8) as well as the new β-oriented derivative (9, 20R). Higher regioselectivity of the acetolysis of 4 was observed using ZnCl2 in acetic anhydride yielding the corresponding 20-α-methyl epoxycholestene 6 in 90% yield. Furthermore, the treatment of each epimeric epoxycholestenes 6 and 9 under basic hydrolysis afforded the new two diastereomeric 23-acetyl-spirostanols 14 and 15 stereospecifically. The structures of all compounds were established using 1D and 2D NMR studies. The assignments of the individual chemical shifts were based on the data for sapogenins and similar compounds previously reported. The stereochemistry of the new spirostanol 15 was confirmed as 20R, 22R, 23S, and 25R by X-ray analysis and also compared with the crystallographic structures of the sarsasapogenin acetate (16), smilagenin (4), and 23-acetyl-sarsasapogenin (17).
Supplemental Material
sj-docx-1-npx-10.1177_1934578X231212341 - Supplemental material for Smilagenin Transformation Products Under Lewis Acid Catalysis in Acetic Anhydride and Synthesis of 23-Acetyl-Spirostanols
Supplemental material, sj-docx-1-npx-10.1177_1934578X231212341 for Smilagenin Transformation Products Under Lewis Acid Catalysis in Acetic Anhydride and Synthesis of 23-Acetyl-Spirostanols by Juan-Pablo García-Merinos, Rebeca Yépez, Claudia M. Ramírez-Lozano, Susana Rincón, Ma. Eugenia Ochoa, Yliana López, Norberto Farfán and Rosa Santillan in Natural Product Communications
Footnotes
Acknowledgements
J.P.G.M. and Y.L. kindly acknowledge to the CIC-UMSNH for financial support. N.F. thanks CONAHCYT, PAIP, PAPIIT (IN200422). C.M.R.L. acknowledges a PhD fellowship from CONAHCYT (No. CVU 957375).
Author Contributions
Juan-Pablo García-Merinos: methodology, investigation, data acquisition, analysis and review & editing. Rebeca Yépez: data acquisition, analysis and review & editing. Claudia M. Ramírez-Lozano: X-ray analysis and writing—review & editing. Susana Rincón: methodology, investigation, validation and review & editing. Ma. Eugenia Ochoa: data acquisition, analysis and review & editing. Yliana López: founding acquisition, conceptualization, validation, writing-review & editing. Norberto Farfán: conceptualization, writing—review & editing, supervision and founding acquisition. Rosa Santillan: supervision, resources, project administration, founding acquisition, writing-original draft, writing—review & editing.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by CIC-UMSNH, CONAHCYT, PAIP and PAPIIT (IN200422).
Ethical Approval
Ethical Approval is not applicable for this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
ORCID iD
Juan-Pablo García-Merinos
Rebeca Yépez
Claudia M. Ramírez-Lozano
Susana Rincón
Ma. Eugenia Ochoa
Yliana López
Norberto Farfán
Rosa Santillan
Supplemental Material
Supplemental material for this article is available online.
HerráizI. Chemical pathways of corticosteroids, industrial synthesis from sapogenins. Microbial Steroids Methods Protocols. 2017;1645:15‐27. https://doi.org/10.1007/978-1-4939-7183-1_2
4.
a. ZhangFMZhangSYTuYQ. Recent progress in the isolation, bioactivity, biosynthesis, and total synthesis of natural spiroketals. Nat Prod Rep. 2018;35(1):75‐104. https://doi.org/10.1039/C7NP00043J b. VeranoALTanDS. Stereocontrolled synthesis of spiroketals: an engine for chemical and biological discovery. Is J Chem. 2017;57(3-4):279‐291. https://doi.org/10.1002/ijch.201600134
5.
a. TobariATeshimaMKoyanagiJ, et al. Spirostanols obtained by cyclization of pseudosaponin derivatives and comparison of anti-platelet agglutination activities of spirostanol glycosides. Eur J Med Chem. 2000;35(5):511‐527. https://doi.org/10.1016/S0223-5234(00)00151-3
b. CallowRKJamesVHT. Epimerization at C25 of steroid sapogenins: sarsasapogenin, neotigogenin and sisalagenin. J Chem Soc. 1955;1671‐1674. https://doi.org/10.1039/JR9550001671 c. WoodwardRBSondheimerFMazurY. The mechanism of the isomerization of steroidal sapogenins at C-25. J Am Chem Soc. 1958;80(24):6693‐6694. https://doi.org/10.1021/ja01557a066
6.
a. GouldDHStaeudleHHershbergEB, etal. Catalytic isomerization of spirostans to furostenols. J Am Chem Soc. 1952;74(14):3685‐3688. https://doi.org/10.1021/ja01134a064
b. UhleFC. C-23 acylation of pseudodiosgenin diacetate. J Org Chem. 1965;30(11):3915‐3920. https://doi.org/10.1021/jo01022a077 c. MarkerREWagnerRBUlshaferPRWittbeckerELGoldsmithDPJRuofCH. Steroidal sapogenins. J Am Chem Soc. 1947;69(9):2167‐2230. https://doi.org/10.1021/ja01201a032 d. WallMESerotaS. Steroidal sapogenins. XX. Configuration of spiroketal side chain at Carbon 22. J Am Chem Soc. 1954;76(10):2850‐2852. https://doi.org/10.1021/ja01639a087 e. WallMESerotaSWitnauerLP. Steroidal sapogenins. XXIV. The hydrochloric acid catalyzed equilibration of 22 ξ,25D- and 22 ξ,25L-spirostanes. J Am Chem Soc. 1955;77(11):3086‐3089. https://doi.org/10.1021/ja01616a045 f. WallMEWalensHA. Steroidal sapogenins. XXVII. Preparation and properties of 20-isosapogenins. J Am Chem Soc. 1955;77(21):5661‐5665. https://doi.org/10.1021/ja01626a058
7.
a. Sandoval-RamírezJCastro-MéndezAMeza-ReyesSReyes-VázquezFSantillánRFarfánN. Preparation of 22,26-epoxycholest-22-ene steroids. Novel transformation of the side chain in sapogenins. Tetrahedron Lett. 1999;40(28):5143‐5146. https://doi.org/10.1016/S0040-4039(99)00884-9
b. RincónSdel RíoRESandoval-RamírezJ A new route for the preparation of the 22,23-dioxocholestane side chain from diosgenin and its application to the stereocontrolled construction of the 22R,23S-diol function. Tetrahedron. 2006;62(11):2594‐2602. https://doi.org/10.1016/j.tet.2005.12.036
c. Meza-ReyesSSandoval-RamírezJMontiel-SmithSβ-Alkoxy-α,β-unsaturated ketone systems in steroidal frameworks and their conversion to 23,24-bisnorcholane lactones. Arkivoc. 2005;vi:307‐320. https://doi.org/10.3998/ark.5550190.0006.626
8.
a. Sandoval-RamírezJMeza-ReyesSDel RíoRE, et al. Regioselective cleavage of rings E and F in sarsasapogenin. Steroids. 2003;68(2):199‐204. https://doi.org/10.1016/S0039-128X(02)00170-8
b. Corona-DíazAGarcía-MerinosJPLópezY Regio- and stereoselective cleavage of steroidal 22-oxo-23-spiroketals catalyzed by BF3·Et2O. Steroids. 2015;100:36‐43. https://doi.org/10.1016/j.steroids.2015.04.004
c. Corona-DíazAGarcía-MerinosJPOchoaMETiCl4 catalyzed cleavage of (25R)-22-oxo-23-spiroketals. Synthesis of sapogenins with furostanol and pyranone E rings on the side chain. Steroids. 2019;152:108488. https://doi.org/10.1016/j.steroids.2019.108488
Viñas-BravoOHernández-LinaresGMata-EsmaMY, et al.1H And 13C NMR of synthetic steroid sapogenins. Part II. C-23 substituted derivatives of (25S)-spirostanes. Arkivoc. 2003;xi:163‐171. https://doi.org/10.3998/ark.5550190.0004.b17
a. EdgingtonPRMcCabePMacraeCF, et al.Mercury: visualization and analysis of crystal structures. J Appl Cryst. 2006;39:453‐457. https://doi.org/10.1107/S002188980600731X
b. FarrugiaLJ. ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI).J Appl Cryst. 1997;30:565‐565. https://doi.org/10.1107/S00218898970 03117
a. AgrawalPKJainDCGuptaRK, etal. Carbon-13 NMR spectroscopy of steroidal sapogenins and steroidal saponins. Phytochemistry. 1985;24(11):2479‐2496. https://doi.org/10.1016/S0031-9422(00)80653-6
b. AgrawalPKBunsawansongPMorrisGA. Complete assignment of the 1H and 13C NMR spectra of steroidal sapogenins: Smilagenin and sarsasapogenin. Magn Reson Chem. 1997;35(7):441‐446. https://doi.org/10.1002/(SICI)1097-458X(199707)35:7<441::AID-OMR104>3.0.CO;2-Q
c. AgrawalPK. Assigning stereodiversity of the 27-Me group of furostane-type steroidal saponins via NMR chemical shifts. Steroids. 2005;70(10):715‐724. https://doi.org/10.1016/j.steroids.2005.04.001
d. AgrawalPK. 25R/25S stereochemistry of spirostane-type steroidal sapogenins and steroidal saponins via chemical shift of geminal protons of ring-F. Magn Reson Chem. 2003;41(11):965‐968. https://doi.org/10.1002/mrc.1278
e. ToriKSeoSTeruiYNishikawaJYasudaF. Carbon-13 NMR spectra of 5β-steroidal sapogenins. Reassignment of the F-ring carbon signals of (25S)-spirostans. Tetrahedron Lett. 1981;22(25):2405‐2408. https://doi.org/10.1016/S0040-4039(01)82920-8
f. ToriKNishikawaJSeoSUomoriAYasudaFKushidaK. 1H NMR spectra of (25S) steroidal sapogenins. Reassignment of the C-20 and C-25 methyl signals. Steroids. 1982;39(1):73‐80. https://doi.org/10.1016/0039-128X(82)90127-1
g. AgrawalPKBunsawansongPMorrisGA. Dependence of the 1H NMR chemical shifts of ring F resonances on the orientation of the 27-methyl group of spirostane-type steroidal sapogenins. Phytochemistry. 1998;47(2):255‐257. https://doi.org/10.1016/S0031-9422(97)00481-0
h. EggertHDjerassiC. 13C NMR spectra of sapogenins. Tetrahedron Lett. 1975;16(42):3635‐3638. https://doi.org/10.1016/S0040-4039(00)91344-3
i. Becerra-MartínezEBañuelos-HernandezAEPérez-HernándezNJoseph-NathanP. Complete 1H NMR assignment of diosgenin benzoate. Nat Prod Commun. 2023;18(7):1‐7. https://doi.org/10.1177/1934578X231188214
j. AgrawalPK. Determining ring-F configuration in spirostane-type steroidal sapogenins by 1H NMR. Indian J Chem. 2005;44B(5):1092‐1094. https://www.researchgate.net/publication/283868802 k. PuriRWongTCPuriRK. Solasodine and diosgenin: 1H and 13C assignments by two-dimensional NMR spectroscopy. Magn Reson Chem. 1993;31(3):278‐282. https://doi.org/10.1002/mrc.1260310313
16.
RosenWEZieglerJBShabicaACShooleryJN. The stereochemistry of steroidal sapogenins. III. N.M.R. Spectra. J Am Chem Soc. 1959;81(7):1687‐1689. https://doi.org/10.1021/ja01516a042
17.
Hernández-LinaresMGSandoval-RamírezJMeza-ReyesSMontiel-SmithSGuerrero-LunaG. Stereospecific synthesis of new steroidal isoxazoles in dry media. Steroids. 2011;76(14):1521‐1526. https://doi.org/10.1016/j.steroids.2011.08.003
18.
a. Rosado-AbonAEsturau-EscofetNFlores-ÁlamoM, etal. The crystal structure of diosgenin acetate and its 23-oxygenated derivatives. J Chem Crystallogr. 2013;43(4):187‐196. https://doi.org/10.1007/s10870-013-0404-3 b. Macías-AlonsoMEsturau-EscofetNFlores-ÁlamoMIglesias-ArteagaMAMoreno-EsparzaR. The crystal structure of 3-Epismilagenin acetate and 23-Oxo-3-epismilagenin acetate
. J Chem Crystallogr. 2011;41:1476‐1482. https://doi.org/10.1007/s10870-011-0126-3
19.
a. SteinerT. The hydrogen bond in the solid state. Angew Chem Int Ed. 2002;41(1):48‐76. https://doi.org/10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U
b. SteinerT. C-H···O Hydrogen bonding in crystals. Crystallogr Rev. 1996;6(1):1‐51. https://doi.org/10.1080/08893119608035394
c. DesirajuGSteinerT. The Weak Hydrogen Bond: In Structural Chemistry and Biology. Oxford University Press, 2001. https://doi.org/10.1093/acprof:oso/9780198509707.001.0001
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.