The ortho-tyrosinol derivative, as a key synthetic intermediate for alkaloidal metabolites of Cordyceps, was synthesized from L-phenylalanine methyl ester via condensation with crotonic acid and oxidative intramolecular lactone formation by using phenyliodine(III) bis(trifluoroacetate). Subsequent elongation of the side chain involving a cross-metathesis reaction and deprotection yielded cordytakaoamide B and cordycepamide C in notably few synthetic steps. Furthermore, the S-configurations at the C-2 position of both cordytakaoamide B and cordycepamide C were confirmed by comparison with the sign of the optical rotation of the synthetic sample.
Entomopathogenic fungi have attracted considerable attention in recent years as a valuable source of biologically active compounds with pharmaceutical utility. In particular, Ophiocordyceps sinensis, formally known as Cordyceps sinensis, is an important species of entomopathogenic fungus that has been widely used in traditional Chinese herbal medicine.1 We recently isolated and structurally characterized 2 novel alkaloid metabolites, cordytakaoamides A (1) and B (2), from the entomopathogenic filamentous fungus Cordyceps takaomontana NBRC 101754.2 The absolute stereochemistry of 1 was determined by total synthesis using L-phenylalanine as starting material, and that of 2 by comparison of the circular dichroic polarization spectrum of 1. In 2020, cordycepamide C (3), which has a similar structure to 2, was isolated from Cordyceps sp. by Zhang et al.3 The absolute configuration of C-2 was determined based on the sign of optical rotation being the same as that of 2 (Figure 1). Here, we report the total synthesis of 2 and 3 based on oxidative ortho-tyrosine synthesis from L-phenylalanine to generate sufficient quantities of samples for biological activity tests and to confirm their absolute configuration at the chiral center.
Structure of cordytakaoamide A (1), B (2), and cordycepamide C (3).
The synthetic strategy was planned according to the previous total synthesis of 12 (Figure 2). Specifically, the diol moiety that is common to 2 and 3 is constructed by reductive cleavage of lactone 4 in the final step. Elongation of the side chain is performed by a cross-metathesis reaction between crotonamide 5 with alkenes bearing appropriate substituents. A key intermediate lactone 5 with an ortho-tyrosine skeleton is constructed by intramolecular regioselective oxidation-lactone formation for the L-phenylalanine derivative 6, which is obtained by acylation and hydrolysis of the L-phenylalanine methyl ester (7).
Retrosynthetic analysis of 2 and 3.
Acylation was performed by treatment of the ester 7 with crotonic acid (8) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) in the presence of a base. Subsequent hydrolysis with LiOH led to an amide 6, whose intramolecular regioselective oxidation was performed using phenyliodine(III) bis(trifluoroacetate) (hereafter PIFA) as an oxidation reagent, as reported by Xue et al.4 After optimizing the reaction conditions, a key intermediate 5 was obtained in 26% yield using excess BF3 etherate. The cross-metathesis reaction for elongation of the side chain of the resulting 5 was carried out in the presence of Grubbs second catalyst5 and alkene 9, prepared according to a previous report,6 under reflux to generate the desired lactone 10 in moderate yield. Finally, the lactone moiety of 10 was reductively cleaved with NaBH4, and cordytakaoamide B (2) was obtained in 66% yield (10% overall yield from the ester 7, Scheme 1). The 1H and 13C nuclear magnetic resonance (NMR) spectra of the synthetic sample were identical to those of natural 2 (Figure 3). Moreover, synthetic sample 2 was found to have an S-configuration at the C-2 position, as shown by the negative optical rotation ([α]D21 −45.4, c 0.30, MeOH). Thus, the absolute configuration of natural (−)-2 (lit. [α]D25 −23.22, c 0.1, MeOH)2 at C-2 was confirmed as S.7
NMR spectra of synthetic and naturally derived 2.
Synthesis of 2.
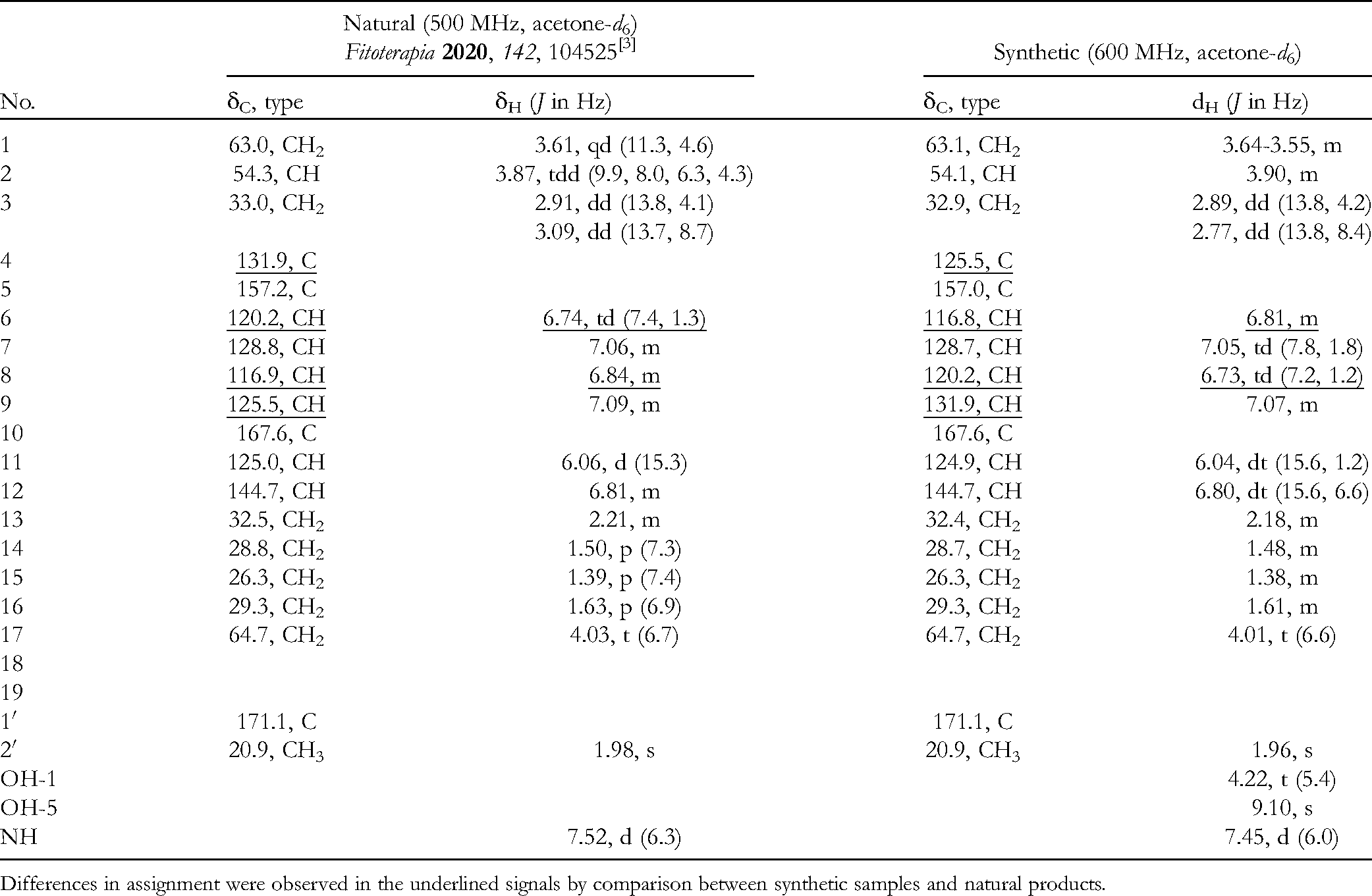
The synthesis of cordycepamide C (3) was initiated from a cross-metathesis reaction for intermediate 5 and alkene 11 prepared by the reduction of hept-6-enoic acid,8 followed by subsequent acetylation. Using the same reaction conditions for cross-metathesis as described earlier, an acetate 12 was obtained with a yield of 59%. After treatment with NaBH4 in EtOH and CH2Cl2,9 cordycepamide C (3) was obtained with 46% yield (Scheme 2, 7.1% yield overall from 7). The 1H and 13C NMR spectra of the synthetic sample were identical to those of natural 3 (Table 1), although some issues were found in the assignment of some signals. After a detailed analysis of the 2D NMR spectra, the original assignments by Zhang et al were revised by replacing the H-6 and H-8, as shown in Table 1.3,10 Synthetic 3 derived from L-phenylalanine having an S-configuration showed a negative optical rotation ([α]D20 −45.1, c 1.35, MeOH). Thus, it was confirmed that the absolute configuration of natural (−)-3 is also S (lit. [α]D −39.59, MeOH).3
Synthesis of 3 from intermediate 5.
Revision of 13C and 1H NMR Spectroscopic Data Assignments Reported by Zhang et al3 by Comparing the Data of Natural and Synthetic 3.
Differences in assignment were observed in the underlined signals by comparison between synthetic samples and natural products.
In conclusion, the synthesis of both cordytakaoamide B (2) and cordycepamide C (3) was achieved from L-phenylalanine methyl ester (7) in 5 steps with an overall yield of 10% and 7.1%, respectively. Moreover, the absolute S-configuration of natural 2 and 3 was confirmed at the C-2 position. In addition, some assignments of the reported NMR spectra for 3 were revised. This synthetic strategy for 2 and 3 is widely applicable to the synthesis of naturally occurring ortho-tyrosine derivatives having variously substituted acyl amide side chains. Furthermore, compounds with similar structures to 2 or 3 that display interesting biological activities, such as glycosyltransferase inhibition and neurotrophic activity, have also been reported.11–14 We are currently studying the synthesis of derivatives of 2 and 3 and evaluating their respective biological activities.
Experimental
General
All reactions sensitive to moisture and/or air were carried out in an atmosphere of argon using dry solvents under anhydrous conditions with oven-dried glassware, unless otherwise noted. Anhydrous dichloromethane (CH2Cl2) was purchased from Kanto Chemical Co. Inc. and used directly without further drying. All other commercial chemicals were of the highest available grade and used directly without additional purification. Analytical thin-layer chromatography was performed using E. Merck silica gel 60 F254 plates (0.25-mm thickness) (Merck). Column chromatography was carried out using Kanto chemical silica gel 60N (40-100 mesh, spherical, neutral). Optical rotations were recorded on a JASCO P-2300 polarimeter (JASCO Corp.), infrared (IR) spectra on a JASCO FT/IR-410 spectrometer, and 1H and 13C NMR spectra on either a Varian 300 or Varian Unity 600 spectrometer (Varian) at room temperature, unless otherwise noted. Chemical shift values are reported in ppm (δ) downfield from tetramethylsilane with reference to internal solvent [1H NMR, CHCl3 (7.26), CHD2OD (3.31), CHD2COCD3 (2.03); 13C NMR, CDCl3 (77.0), CD3OD (49.0), CD3COCD3 (206.3)], unless otherwise noted. Coupling constants (J) are reported in Hertz (Hz). The following abbreviations are used to designate the multiplicities: s = singlet; d = doublet; t = triplet; m = multiplet; br = broad. Chemical ionization (CI) mass spectra were measured on a JEOL MStation JMS-700.
(E)-but-2-enoyl-l-phenylalanine (6)
Crotonic acid (442.6 mg, 5.141 mmol) was added to a solution of L-phenylalanine methyl ester (7) (1.01 g, 0.605 mmol), EDCI (985.5 mg, 5.141 mmol), and iPr2NEt (1.84 mL, 10.283 mmol) in CH2Cl2 (9.4 mL) at room temperature. The resultant solution was stirred at room temperature for 7 h before the reaction was quenched with 1 M aq HCl. The resultant solution was extracted with EtOAc and the organic layer was dried over Na2SO4, filtered, and then concentrated under reduced pressure. Purification of the residue by column chromatography (silica gel, hexane/EtOAc = 5/1 to 2/1) gave the amide as a colorless oil. The resulting methyl ester was dissolved in MeOH/H2O (4:1, 41.1 mL) before the addition of LiOH·H2O (379.6 mg, 9.046 mmol) at room temperature. After stirring for 2 h at room temperature, the reaction was quenched with 1 M aq HCl. The resultant solution was extracted with EtOAc and the organic layer was dried over Na2SO4, filtered, and then concentrated under reduced pressure to furnish carboxylic acid 6 (1.087 g, 4.660 mmol) as a white solid: [α]D22 + 21.8 (c 0.99, MeOH); IR (film) 3338, 3030, 2916, 1730, 1668, 1621, 1538, 1455, 1216 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.27-7.19 (m, 5H), 6.74 (dq, J = 15.2, 6.9 Hz, 1H), 5.95 (dq, J = 15.2, 1.7 Hz, 1H), 4.72 (dd, J = 8.8, 5.0 Hz, 1H), 3.22 (dd, J = 13.8, 5.0 Hz, 1H), 2.98 (dd, J = 13.8, 8.8 Hz, 1H), 1.82 (dd, J = 6.9, 1.7 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 173.7, 167.3, 140.5, 137.4, 129.2 (2C), 128.4 (2C), 126.8, 124.6, 54.0, 37.4, 16.8; high-resolution mass spectrometry (HRMS) (CI) calcd for C13H16NO3 [(M)+] 234.1125, found 234.1130.
(S,E)-N-(2-oxochroman-3-yl)but-2-enamide (5)
To a solution of carboxylic acid 6 (369.8 mg, 1.585 mmol) and PIFA (749.8 mg, 1.744 mmol) in CH2Cl2 (15.8 mL) at room temperature was added BF3·OEt2 (1.99 mL, 15.85 mmol). After stirring for 3 h at room temperature, the mixture was diluted with CH2Cl2 and the resulting solution was washed successively with saturated aqueous NaHCO3 and saturated aqueous NaCl solution, dried over Na2SO4, filtered, and then concentrated under reduced pressure. Purification of the residue by column chromatography (silica gel, hexane/EtOAc = 1/0 to 5/1) gave lactone 5 (96.7 mg, 0.418 mmol) as a colorless oil: [α]D21 + 49.4 (c 0.50, CHCl3); IR (film) 3287, 3048, 2967, 2915, 1767, 1669, 1630, 1539, 1487, 1457, 1368, 1230, 1156 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.28 (m, 1H), 7.21 (m, 1H), 7.13 (ddd, J = 7.4, 1.1, 1.1 Hz, 1H), 7.06 (dd, J = 8.2, 0.8 Hz, 1H), 6.93 (dq, J = 15.1, 6.9 Hz, 1H), 6.68 (brd, J = 5.8 Hz, 1H), 5.94 (dq, J = 15.1, 1.7 Hz, 1H), 4.87 (dt, J = 13.7, 6.6 Hz, 1H), 3.51 (dd, J = 15.4, 6.6 Hz, 1H), 2.95 (m, 1H), 1.88 (dd, J = 6.9, 1.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 168.8, 165.8, 150.7, 141.5, 128.7, 128.6, 125.0, 124.1, 121.3, 116.6, 48.1, 30.4, 17.8; HRMS (CI) calcd for C13H14NO3 [(M + H)+] 232.0968, found 232.0968.
To a solution of alkene 5 (35.6 mg, 0.153 mmol) and alkene 11 (24.0 mg, 0.153 mmol) in CH2Cl2 (1.5 mL) at room temperature was added Grubbs second catalyst (6.5 mg, 7.6 μmol). The resultant solution was refluxed for 18 h before being concentrated under reduced pressure. Purification of the residue by column chromatography (silica gel, hexane/acetone = 4/1 to 1/1) gave lactone 12 (31.1 mg, 0.0898 mmol) as a white solid: [α]D21 + 34.6 (c 1.02, CHCl3); IR (film) 3288, 3048, 2932, 2856, 1758, 1667, 1627, 1541, 1369, 1234 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.31-7.05 (m, 4H), 6.91 (dt, J = 15.2, 7.1 Hz, 1H), 6.69 (d, J = 5.9 Hz, 1H), 5.92 (d, J = 15.2 Hz, 1H), 4.86 (ddd, J = 13.8, 6.4, 6.4 Hz, 1H), 4.06 (t, J = 6.7 Hz, 2H), 3.52 (dd, J = 15.2, 6.4 Hz, 1H), 2.96 (dd, J = 15.2, 13.8 Hz, 1H), 2.22 (q, J = 6.8 Hz, 2H), 2.06 (s, 3H), 1.69-1.35 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 171.2, 168.6, 165.8, 150.7, 145.9, 128.7, 128.6, 125.0, 122.8, 121.3, 116.6, 64.2, 48.2, 31.8, 30.4, 28.3, 27.6, 25.4, 20.9; HRMS (CI) calcd for C19H24NO5 [(M + H)+] 346.1649, found 346.1661.
Cordycepamide C (3)
To a solution of lactone 12 (70.0 mg, 0.204 mmol) in EtOH/CH2Cl2 (1:1, 2 mL) at 0 °C was added NaBH4 (15.4 mg, 0.408 mmol). The resultant solution was stirred at 0 °C for 2 h before being diluted with EtOAc, followed by 1 M aq HCl. The organic layer was separated, dried over Na2SO4, filtered, and then concentrated under reduced pressure. Purification of the residue by column chromatography (silica gel, hexane/EtOAc = 5/1 to 1/1) gave cordycepamide C (3) (32.6 mg, 0.0938 mmol) as a colorless oil: [α]D20 −45.1 (c 1.35, MeOH); IR (film) 3328, 2932, 2858, 1732, 1718, 1666, 1624, 1541, 1456, 1364, 1242 cm−1; 1H NMR (600 MHz, acetone-d6) δ 9.10 (s, 1H), 7.45 (d, J = 6.0 Hz, 1H), 7.07 (m, 1H), 7.05 (td, J = 7.2, 1.2 Hz, 1H), 6.81 (m, 1H), 6.80 (dt, J = 15.6, 6.6 Hz, 1H), 6.73 (td, J = 7.2, 1.2 Hz, 1H), 6.04 (dt, J = 15.6, 1.2 Hz, 1H), 4.22 (t, J = 5.4 Hz, 1H), 4.01 (t, J = 6.6 Hz, 2H), 3.90 (m, 1H), 3.64-3.55 (m, 2H), 2.89 (dd, J = 13.8, 4.2 Hz, 1H), 2.77 (dd, J = 13.8, 8.4 Hz, 1H), 2.18 (m, 2H), 1.96 (s, 3H), 1.61 (m, 2H), 1.48 (m, 2H), 1.38 (m, 2H); 13C NMR (150 MHz, acetone-d6) δ 171.1, 167.6, 157.0, 144.7, 131.9, 128.7, 125.5, 124.9, 120.2, 116.8, 64.5, 63.1, 54.1, 32.9, 32.4, 29.3, 28.7, 26.3, 20.9; HRMS (CI) calcd for C19H28NO5 [(M + H)+] 350.1962, found 350.1968.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221099141 - Supplemental material for Short-step Synthesis of Cordytakaoamide B and Cordycepamide C via Intramolecular Regioselective Oxidation-Lactone Formation
Supplemental material, sj-docx-1-npx-10.1177_1934578X221099141 for Short-step Synthesis of Cordytakaoamide B and Cordycepamide C via Intramolecular Regioselective Oxidation-Lactone Formation by Yusuke Kasai, Takamitsu Ogawa, Mao Kawata, Kana Tanigawa, Abdelsamed I. Elshamy, Tatsuro Yoneyama, Masaaki Noji, Akemi Umeyama and Hiroshi Imagawa in Natural Product Communications
Footnotes
Acknowledgments
We thank Dr Katsuyuki Nakashima and Dr Yasuko Okamoto (Center for Instrumental Analysis, Tokushima Bunri University) for performing the 600 MHz NMR spectra and mass spectrometry measurements, respectively.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical Approval is not applicable for this article.
ORCID iDs
Yusuke Kasai
Tatsuro Yoneyama
Hiroshi Imagawa
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Supplemental material
Supplemental material for this article is available online.
References
1.
LoH-CHsiehCLinF-YHsuT-H. A systematic review of the mysterious caterpillar fungus Ophiocordyceps sinensis in DongChongXiaCao. J Tradit Complement Med. 2013;3(1):16‐32. doi:10.1016/S2225-4110(16)30164-X
2.
HamaMElshamyAIYoneyamaT, et al.New alkaloidal metabolites from cultures of entomopathogenic fungus Cordyceps takaomontana NBRC 101754. Fitoterapia. 2019;139:104364. doi: 10.1016/j.fitote.2019.104364
3.
FanWLiERenJWangWLiuXZhangY. Cordycepamides A−E and cordyglycoside A, new alkaloidal and glycoside metabolites from the entomopathogenic fungus Cordyceps sp. Fitoterapia. 2020;142:104525. doi:10.1016/j.fitote.2020.104525
4.
GuYXueK. Direct oxidative cyclization of 3-arylpropionic acids using PIFA or oxone: synthesis of 3,4-dihydrocoumarins. Tetrahedron Lett. 2010;51(1):192‐193. doi:10.1016/j.tetlet.2009.10.112
The large difference in the optical rotation values between synthetic and natural 2 can be attributed to the lower purity of the naturally derived sample. This is suggested by the fact that a pure synthetic sample of 3, which has a similar structure to 2, shows an absolute value of optical rotation that is about the same as the synthetic sample of 2.
8.
Hept-6-enoic acid, whose synthesis was reported in our previous paper2 was reduced with LiAlH4 in Et2O. And the resulting alcohol was treated with acetic anhydride to give alkene 11.
9.
El AssalMPeixotoPACoffinierR, et al.Synthesis of scyphostatin analogues through hypervalent iodine-mediated phenol dearomatization. J Org Chem. 2017;82(22):11816‐11828. doi:10.1021/acs.joc.7b02366
10.
The assignments for C-4 and C-9 were also revised by replacing each other.
11.
WarrenKRMisraRSAroraRCRadinNS. Glycosyltransferases of rat brain that make cerebrosides: substrate specificity, inhibitors, and abnormal products. J Neurochem. 1976;26(6):1063‐1072. doi:10.1111/j.1471-4159.1976.tb06987.x
12.
ChengYSchneiderBRieseUSchubertBLiZHamburgerM. Farinosones A–C, Neurotrophic alkaloidal metabolites from the entomogenous deuteromycete Paecilomyces fainosus. J Nat Prod. 2004;67(11):1854‐1858. doi:10.1021/np049761w
13.
JessenHBarbaras DJHamburgerMGademannK. Total synthesis and neuritotrophic activity of farinosone C and derivatives. Org Lett. 2009;11(15):3446‐3449. doi:10.1021/ol901277q
14.
BurchPChiccaAGertschJGademannK. Functionally optimized neurogenic farinosone C analogs: SAR-study and investigations on their mode of action. ACS Med Chem Lett. 2014;5(2):172‐177. doi:10.1021/ml400435h
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.