Seven new dihydrobenzofurans and 2 new propynyl thiophenes were isolated from the roots of Eupatorium heterophyllum together with 13 known compounds. The compounds were characterized using spectroscopic methods including 2D NMR, infrared, and mass spectrometric techniques. Aerial parts of this plant have been known to contain various sesquiterpenoids and displayed high chemical diversity (several compounds isolated and/or identified) among their chemical constituents depending on the collection site. Nevertheless, we found that the chemical diversity in the roots was lower than in the aerial parts.
Eupatorium species (Asteraceae) are herbaceous perennials, which are widely distributed worldwide, especially in East Asia.1 Sesquiterpenoids with germacrane, guaiane, eudesmane skeletons have been isolated from this genus. Interestingly, our studies demonstrated that plant samples of Eupatorium species, which were collected from different locations in Japan showed significant variations in their chemical compositions.2–4 The differences in chemical compositions were primarily observed in the above-ground parts.5 Additionally, an investigation on the roots (underground part) of E. heterophyllum, which were collected from five districts in China showed only small differences in chemical compositions.6 In this study, we further examined the roots of E. heterophyllum, which were collected from Sichuan, China. The isolation, determination of structures, and identification of new dihydrobenzofurans and propynylthiophenes were described in this paper.
Results and Discussion
E. heterophyllum plant samples were collected from Sichuan, China. The dried roots of the plant were extracted with methanol (MeOH), and the extracts were fractionated by silica gel column chromatography. The separation of the fractions via preparative high-performance liquid chromatography (HPLC) using various types of columns afforded seven new compounds (1 – 7), an inseparable mixture of diastereomeric isomers of new compounds (8 and 9), along with 13 known compounds. By comparison of spectroscopic data with data in literatures, the known compounds were identified as six dihydrobenzofurans (10,711,812,913,614,10,11 and 1512), two benzofurans (167,13 and 1714), one enyne-type compound (186), one dihydroxy-formyl-acetophenone (1915), and three propynyl thiophenes (20,1621,16 and 226) (Figure 1).
Structures of new (1-9) and known (10-22) compounds isolated from the dried roots of E. heterophyllum (Sichuan, China).
Compound 1 was isolated as a yellow amorphous powder, and its molecular formula C13H14O4 was determined based on the molecular ion (M+) peak at m/z 234.0891 in the high-resolution chemical ionization mass spectrometry (HR-CI-MS) (calculated molecular mass for C13H14O4 = 234.0892). The infrared (IR) spectrum showed the presence of hydroxyl (3390 cm−1) and conjugated carbonyl groups (1643 cm−1). The 1H and 13C NMR spectroscopic data (Table 1) showed signals attributable to an acetyl [δH 2.84 (s, H3-2′), δC 204.2 (C-1′)], an allylic methyl [δH 1.75 (s, H3-10), δC 17.7 (C-10)], mutually ortho-coupled aromatic protons [δH 7.10 (d, J = 8.8 Hz, H-7); 6.97 (d, J = 8.8 Hz, H-6)], and an exomethylene group [δH 5.06 (d, J = 0.7 Hz, H-9a), 4.92 (d, J = 0.7 Hz, H-9b); δC 112.7 (C-9)]. Additionally, the hydroxyl proton signal at δH 12.25 (s, 5-OH) suggested the formation of a hydrogen bond between a hydroxyl group and a carbonyl group. The 1H–1H homonuclear correlated spectroscopy (COSY) and proton-detected heteronuclear multiple bond correlation (HMBC) spectra (Figure 2) indicated that 1 is a benzofuran-type compound with two hydroxyl groups at C-3 and C-5 and an acetyl group at C-4. The relative stereochemistry of C-2 and C-3 was found to have a trans geometry on the basis of the nuclear overhauser effect between C-3-OH and H-2 and between H-3 and allylic methyl (H-10). This geometry was supported by the small JH−2−H−3 (1.7 Hz) observed.17,18 Based on these spectroscopic results, 1 was determined to be 4-acetyl-3β,5-dihydroxy-2α-(propen-2-yl)-2,3-dihydrobenzofuran. The absolute configuration of 1 was not determined.
Key 2D NMR correlations for compound 1.
1H (500 MHz) and 13C (126 MHz) NMR Data of Compounds 1-3.
Position
1
2
3
δH
(mult., J in Hz)
δC
δH
(mult., J in Hz)
δC
δH
(mult., J in Hz)
δC
2
4.95
(1H, br s)
93.2
4.99
(1H, d, 1.2)
90.7
4.85
(1H, d, 4.6)
93.5
3
5.31
(1H, dd, 9.6, 1.7)
78.0
6.38
(1H, d, 1.2)
78.8
5.13
(1H, dd, 7.6, 4.6)
76.6
3a
–
–
124.9
–
–
120.3
–
–
137.4
4
–
–
116.6
–
–
116.7
7.01
(1H, s)
114.9
5
–
–
158.1
–
–
158.5
–
–
157.7
6
6.97
(1H, d, 8.8)
122.0
7.05
(1H, d, 9.0)
122.6
–
–
120.0
7
7.10
(1H, d, 8.8)
119.3
7.17
(1H, d, 9.0)
119.3
7.18
(1H, s)
109.6
7a
–
–
153.2
–
–
154.8
–
–
151.9
8
–
–
141.0
–
–
140.2
–
–
141.2
9
5.06
(1H, d, 0.7)
112.7
5.07
(1H, d, 1.0)
113.5
5.12
(1H, d, 0.5)
113.0
4.92
(1H, d, 0.7)
4.98
(1H, d, 1.0)
4.96
(1H, d, 0.5)
10
1.75
(3H, s)
17.7
1.87
(3H, s)
18.4
1.77
(3H, s)
17.5
1’
–
–
204.2
–
–
203.2
–
–
204.0
2’
2.84
(3H, s)
30.4
2.55
(3H, s)
30.1
2.61
(3H, s)
27.0
3-OH
2.32
(1H, d, 9.6)
–
2.25
(1H, d, 7.6)
–
3-OCH3
5-OH
12.25
(1H, s)
–
12.42
(1H, s)
12.05
(1H, s)
–
1”
–
–
167.2
2”
–
–
126.8
3”
6.18
(1H, qq, 7.1, 1.5)
140.6
4”
1.96
(3H, dq, 7.1, 1.5)
16.0
5”
1.87
(3H, quint, 1.5)
20.5
Abbreviation: Measured in CDCl3.
Compound 2 was obtained as a yellow amorphous powder. The molecular formula was determined to be C18H20O5 via HR-CI-MS. The 1H and 13C NMR spectra of 2 (Table 1) resembled those of 1, suggesting that 2 is also a benzofuran derivative that is related to 1. Comparison of the NMR signals of 1 and 2 showed that the spectra of 2 showed additional signals arising from an angeloyl ester group [δH 6.18 (qq, J = 7.1,1.5 Hz, H-3″); 1.96 (dq, J = 7.1,1.5 Hz, H-4″); 1.87 (quint, J = 1.5 Hz, H-5″), δC 167.2 (C-1″), 126.8 (C-2″), 140.6 (C-3″), 16.0 (C-4″), 20.5 (C-5″)]. This was accompanied by a large low field shift of H-3 to δH 6.38 (Δδ 1.07) in the 1H NMR spectrum. Relative stereochemistry was the same for 1 and 2 based on the NOESY correlations (Figure 3). These observations indicated that 2 is 4-acetyl-3β-angeloyloxy-5-hydroxy-2α-(propen-2-yl)-2,3-dihydrobenzofuran. This compound is a positional isomer of the known compound 11 (Figure 1).
Key 2D NMR correlations for compound 2.
The molecular formula of 3 was found to be the same as that of 1 by HR-CI-MS. The 1H and 13C NMR results (Table 1) obtained for 3 and 1 suggested a benzofuran skeleton for both molecules; thus, we concluded that 3 and 1 are isomers. However, two aromatic protons in 3 appeared as singlet signals [δH 7.18 (s, H-7); 7.01 (s, H-4)], and 1H–1H COSY, HMBC, and NOESY correlations (Figure 4) indicated that the location of the hydroxyl and acetyl groups in 3 were in C-5 and C-6, respectively. The HMBC and NOESY correlations also confirmed that the structure of the furan ring of 3 is the same as that of 1. Accordingly, 3 was identified as 6-acetyl-3β,5-dihydroxy-2α-(propen-2-yl)-2,3-dihydrobenzofuran.
Key 2D NMR correlations for compound 3.
The 1H and 13C NMR spectra of 4 were closely related to known compound 12 (Figure 1). The HR-CI-MS analyses of 4 and 12 suggested a molecular weight of 248 amu for both compounds. The difference was found in the 1H NMR signals of their furan ring part, where the JH−2−H−3 values of 4 and 12 were 6.1 and 2 Hz,9 respectively (Table 2). After considering the HMBC and NOESY correlations shown in Figure 5, 4 was determined to be 5-acetyl-6-hydroxy-3α-methoxyl-2α-(propen-2-yl)-2,3-dihydrobenzofuran.
Key 2D NMR correlations for compound 4.
1H (500 MHz) and 13C (126 MHz) NMR Data of Compounds 4-6.
Position
4
5
6
δH
(mult., J in Hz)
δC
δH
(mult., J in Hz)
δC
δH
(mult., J in Hz)
δC
2
5.01
(1H, d, 6.1)
90.6
4.56
(1H, d, 7.0)
89.0
4.79
(1H, d, 5.9)
85.8
3
4.82
(1H, d, 6.1)
79.5
6.40
(1H, d, 7.0)
72.0
6.31
(1H, d, 5.9)
71.9
3a
–
–
118.8
–
–
117.5
–
–
117.5
4
7.74
(1H, s)
128.8
7.85
(1H, s)
130.1
7.94
(1H, s)
130.7
5
–
–
114.2
–
–
114.9
–
–
114.9
6
–
–
167.0
–
–
167.1
–
–
167.1
7
6.46
(1H, s)
99.1
6.46
(1H, s)
99.0
6.46
(1H, s)
99.2
7a
–
–
166.5
–
–
166.3
–
–
166.9
8
–
–
139.3
–
–
54.9
–
–
54.9
9
5.26
(1H, d, 0.7)
113.9
2.90
(1H, d, 4.6)
50.4
3.12
(1H, d, 4.6)
51.2
5.13
(1H, q, 1.5)
2.71
(1H, d, 4.6)
2.77
(1H, d, 4.6)
10
1.89
(3H, s)
19.3
1.45
(3H, s)
17.5
1.48
(3H, s)
18.8
1’
–
–
202.4
–
–
202.9
–
–
202.9
2’
2.59
(3H, s)
26.3
2.56
(3H, s)
26.4
2.57
(3H, s)
26.5
3-OH
3-OCH3
3.35
(3H, s)
56.5
5-OH
6-OH
13.01
(1H, s)
–
13.01
(1H, s)
–
13.01
(1H, s)
–
1”
–
–
167.2
–
–
167.2
2”
–
–
126.8
–
–
126.8
3”
6.18
(1H, qq, 7.1, 1.4)
140.7
6.18
(1H, qq, 7.1, 1.4)
140.7
4”
2.04
(3H, dq, 7.1, 1.4)
15.9
2.01
(3H, dq, 7.1, 1.4)
15.9
5”
1.90
(3H, m)
20.5
1.84
(3H, m)
20.4
Abbreviation: Measured in CDCl3.
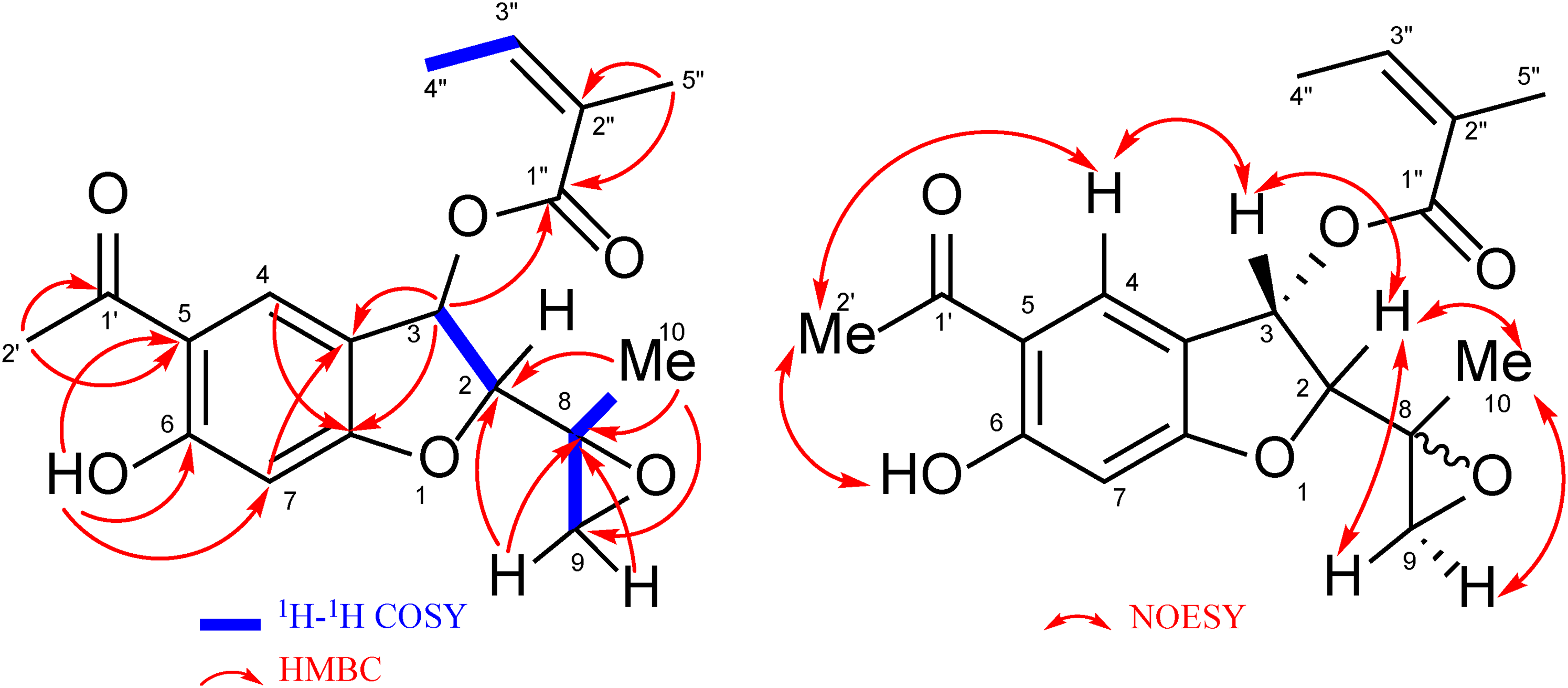
Compounds 5 and 6 were obtained as an inseparable mixture (detected in a ratio of 3:1 [5:6] based on the integration of 1H NMR signals). The CI-MS of the mixture exhibited one [M + H]+ peak at m/z 333, and the HR-CI-MS confirmed a molecular formula of C18H20O6, which was 16 amu larger than that found for 10 and 2. The 1H NMR signals obtained from 5 and 6 were distinguishable (Table 2), and their chemical shifts and HMBC correlations (Figure 6) suggested structural similarities of these compounds to 10. Nevertheless, instead of the olefinic carbons of C-8 and C-9 found in 10, signals attributable to oxymethylene protons were observed [5: δH 2.90 (d, J = 4.6 Hz, H-9a); 2.71 (d, J = 4.6 Hz, H-9b), 6: δH 3.12 (d, J = 4.6 Hz, H-9a); 2.77 (d, J = 4.6 Hz, H-9b)]. Additionally, the C-10 methyl protons were shifted to the upper field (Δδ - 0.4 compared with that of 10). Considering the unsaturation index of 9 for both 5 and 6, these compounds were decided to be a pair of diastereomeric isomers of epoxides of 10 as shown in Figure 6. The chemical shifts of C-8 and C-9 supported the formation of epoxides at these positions (Table 2). Thus, 5 and 6 were characterized as C-8 epimers of 5-acetyl-3α-angeloyloxy-6-hydroxy-2α-(2-methyloxiran-2-yl)-2,3-dihydrobenzofuran.
Key 2D correlations for compounds 5 and 6.
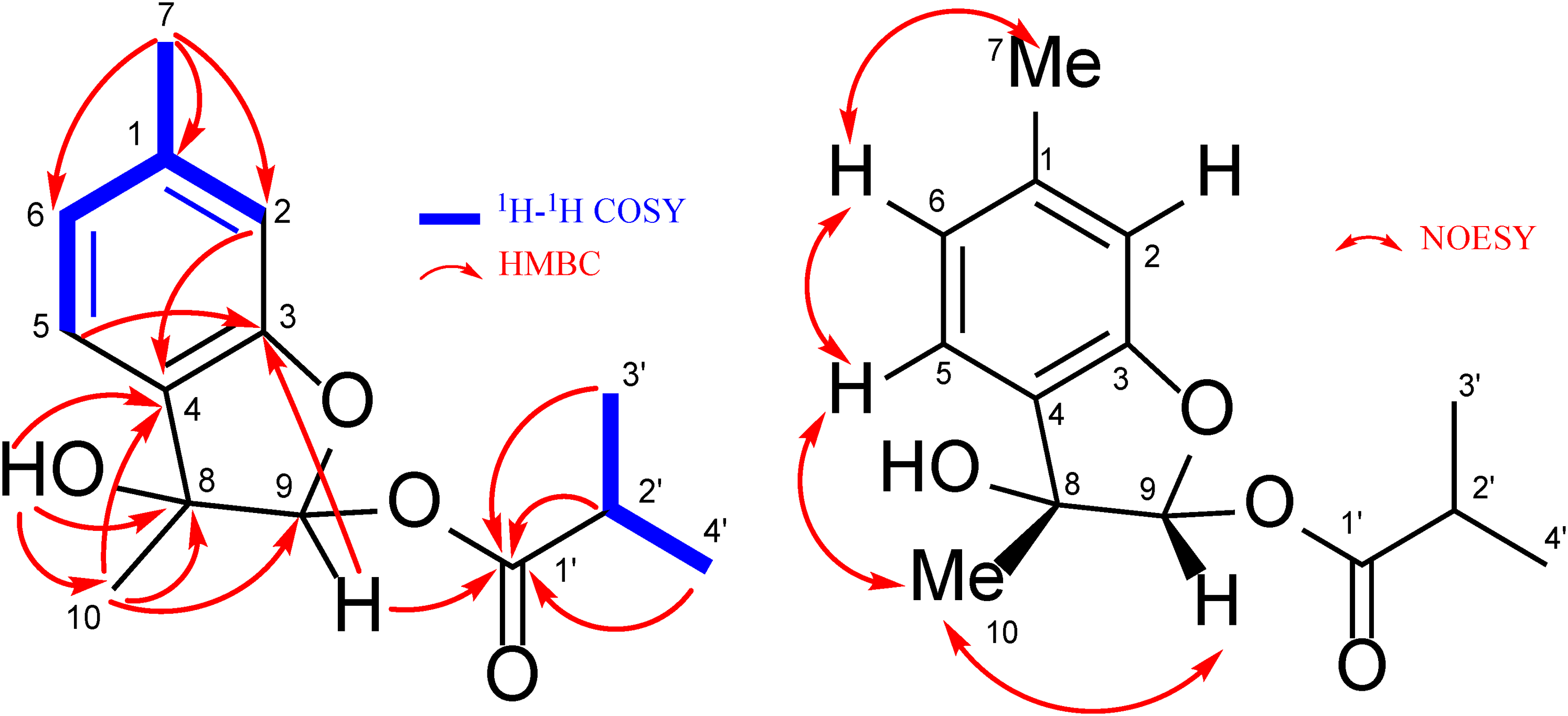
Compound 7 was isolated as a colorless amorphous powder. The molecular formula corresponded to C14H18O4 based on HR-CI-MS. The 1H NMR spectrum (Table 3) showed signals due to a 12,4-trisubstituted aromatic ring [δH 6.76 (s, H-2), 7.22 (d, J = 7.6 Hz, H-5), 6.85 (dd, J = 7.6, 0.5 Hz, H-6)] and an isobutanoyl group [δH 2.59 (sept, J = 7.1 Hz, H-2′), 1.19 (d, J = 7.1 Hz, H3-3′), 1.18 (d, J = 7.1 Hz, H3-4′)]. The presence of an ester moiety was supported by the strong absorption at 1747 cm−1 in the IR spectrum. The presence of an acetal methine carbon [δH 6.50 (s, H-9), δc 104.9 (C-9)] was shown by 13C NMR and heteronuclear single quantum coherence spectra. The large chemical shift value of the C-9 methine proton indicated the esterification of an acetal hydroxy group. In the HMBC spectrum, correlations from H3 to 7 (δH 2.35, s) to C-1 (δc 141.6), C-2 (δc 111.7) and C-6 (δc 123.1), and from H3 to 10 (δH 1.62, s) to C-4 (δc 127.6), C-8 (δc 79.8) and C-9 (δc 104.9) revealed the presence of a menthane skeleton in 7. Furthermore, the HMBC correlation of H-9 acetal proton with C-3 oxygenated aromatic carbon (δc 158.5) and C-1′ ester carbonyl carbon (δc 175.7) suggested a planar structure of 7 (Figure 7). As for relative stereochemistry, the NOESY correlation between H-9 and H3 to 10 indicated that these protons were located at the same side of the molecule. Based on these results, 7 was identified as 3,9β-epoxy-9α-isobutanoyloxymentha-13,5-trien-8α-ol.
Key 2D NMR correlations for compound 7.
1H (500 MHz) and 13C (126 MHz) NMR Data of 7.
δH
(mult., J in Hz)
δC
1
–
–
141.6
2
6.76
(1H, s)
111.7
3
–
–
158.5
4
–
–
127.6
5
7.22
(1H, d, 7.6)
122.7
6
6.85
(1H, dd, 7.6, 0.5)
123.1
7
2.35
(3H, s)
21.7
8
–
–
79.8
9
6.50
(1H, s)
104.9
10
1.62
(3H, s)
20.1
1’
–
–
175.7
2’
2.59
(1H, sept, 7.1)
34.1
3’
1.19
(3H, d, 7.1)
18.8
4’
1.18
(3H, d, 7.1)
18.5
8-OH
2.03
(1H, s)
–
Abbreviation: Measured in CDCl3.
1H and 13C NMR data of 8 and 9 were closely related to those of known compounds 21 and 22 (Figure 1), respectively. This suggests that 8 and 9 are propyn-1-yl thiophene derivatives. The presence of a triple bond in each compound was confirmed by absorptions (8: 2229 cm−1; 9: 2229 cm−1) in the IR spectra and quaternary carbon signals [8: δC 73.1 (C-6), 96.2 (C-7); 9: δC 73.2 (C-6), 94.8 (C-7)] in the 13C NMR spectra (Table 4). The differences in the NMR spectra of 8 and 21 were the appearance of the signals attributable to a hydroxymethyl group in 8 [δH 4.63 (d, J = 4.7 Hz, H2-2′), 3.55 (t, J = 4.7 Hz, 2′-OH); δC 67.8 (C-2′)] instead of the signals of C-2’ methyl group in 21. Thus, 8 was given the structure shown in Figure 8 and was identified as 2-(hydroxyacetyl)-3-methoxy-5-(propyn-1-yl)thiophene. The HMBC and NOESY correlations illustrated in Figure 8 were consistent with the proposed structure.
Key 2D NMR correlations for compound 8.
1H (500 MHz) and 13C (126 MHz) NMR Data of Compounds 8 and 9.
Compared with the NMR spectra of 22, 9 showed additional signals from a malonate group [δH 3.87 (d, J = 15.7 Hz, H-2‴a), 3.82 (d, J = 15.7 Hz, H-2‴b); δC 42.0 (C-2‴), 167.3 (C-1‴), 169.0 (C-3‴)] and large low field shifts of glucose H2 to 6 [δH 5.16 (dd, J = 11.6, 1.9 Hz, H-6a″), 4.80 (dd, J = 11.6, 7.1 Hz, H-6b″)] (Table 4). HMBC correlations between glucose H2 to 6 and malonate H2 to 2‴ to the same ester carbonyl carbon at δC 167.3 (C-1‴) confirmed the esterification of the malonyl group to glucose C-6 position (Figure 9). Based on these observations, 9 was identified as 2-acetyl-3-hydroxy-5-(propyn-1-yl)thiophene 3-O-(6-O-malonyl)-β-glucoside.
Key 2D NMR correlations for compound 9.
This study isolated and identified nine new and 13 known compounds in the roots of E. heterophyllum. These compounds mainly belonged to benzofurans, dihydrobenzofurans, and propynyl thiophenes. The major constituents of the sample were a benzofuran (16) and two dihydrobenzofurans (10 and 12). This finding was consistent with a similar previous study.6 Interestingly, the aerial parts of this plant have been shown to contain mainly sesquiterpenoids and displayed considerable chemical diversity, which was dependent on the collection site.5 However, our results clearly suggested a lower chemical diversity in the roots.6 The difference in the chemical diversity between the aerial parts and roots may be important for plant physiology and chemical ecology. For example, biosynthesis in the aerial part was most likely sensitive to changes in the environmental conditions, whereas biosynthesis in the roots was generally not affected by environmental factors. Our results also raise the question of why the root and aerial parts of the studied plant show different biosynthetic systems.
Experimental
General
The IR spectra were obtained using an FT/IR-400 spectrophotometer (JASCO, Tokyo, Japan). The 1H, 13C, and 2D NMR spectra were measured using a Varian Unity plus 500 spectrometer (1H: 500 MHz; 13C: 126 MHz), JEOL JNM-AL 400 spectrometer (1H: 400 MHz; 13C: 100 MHz). The mass spectra were obtained with JEOL JMS-700N mass spectrometer. Preparative HPLC columns used were COSMOSIL 5C18-PAQ (20 mm i.d. × 250 mm), COSMOSIL 5SL-II (4.6 mm i.d. × 250 mm), and COSMOSIL 5SL-II (10 mm i.d. × 250 mm) from Nacalai Tesque Inc. (Kyoto, Japan); Inertsil CN-3 (4.6 mm i.d. × 250 mm) from GL Science (Tokyo, Japan); and YMC-Pack Diol-120-NP (4.6 mm i.d. × 250 mm) from YMC Co., Ltd (Kyoto, Japan). Preparative HPLC was with a JASCO PU2080 pump system. Silica gel 60N (100-210 μm) (Kanto Chemical Co., Inc., Tokyo, Japan) was used for column chromatography. TLC was with silica gel 60 F254 plates (Merck).
Plant Materials
The plant materials were collected in Maoxian, Sichuan, China during August 2015. A voucher specimen (#2015-25) was deposited at the herbarium of Kunming Institute of Botany. The plant was identified by Dr Takayuki Kawahara, Japan Forest Technology Association, General Incorporated Association, Japan.
Extraction and Isolation
The dried root (122.5 g) of E. heterophyllum was extracted twice with MeOH at room temperature. The extract (16.0 g) was separated by silica gel column chromatography (50 mm i.d. × 250 mm, n-hexane:CHCl3 = 9:1, 7:3, 1:1, CHCl3:MeOH = 1:0, 98:2, 95:5, 9:1, 7:3, 0:1) into nine fractions: Fr. A–I. Fr. C (344 mg) was separated by preparative HPLC using COSMOSIL 5SL-II (10 mm i.d. × 250 mm, 6 ml/min, n-hexane:EtOAc = 95:5) to yield 20 (44.7 mg) and 16 (166.2 mg). Fr. D (389 mg) was separated into six subfractions by silica gel column chromatography (20 mm i.d. × 150 mm, n-hexane:EtOAc = 1:0, 99:1, 98:2, 96:4, 94:6, 9:1, 8:2, EtOAc, MeOH). Fr. D-3 (259 mg) was identified as 10. Fr. D-2 (82.7 mg) was separated by COSMOSIL 5SL-II (10 mm i.d. × 250 mm, 6 ml/min, n-hexane:EtOAc = 9:1) to give Fr. D-2 to 1 (26.0 mg), 11 (33.1 mg), and 16 (13.2 mg). Separations of the Fr. D-2 to 1 were done via successive preparative HPLC using YMC-Pack Diol-120-NP (4.6 mm i.d. × 250 mm, 2 ml/min, n-hexane:EtOAc = 95:5), YMC-Pack Diol-120-NP (4.6 × 250 mm, 1 ml/min, n-hexane:EtOAc = 99:1), and Inertsil CN-3 (4.6 mm i.d. × 250 mm, 1 ml/min, n-hexane:EtOAc = 99.5:0.5). These afforded 14 (0.8 mg), 15 (1.9 mg), and 2 (0.6 mg). Fr. D-4 (19.1 mg) was separated by preparative HPLC using COSMOSIL 5SL-II (4.6 mm i.d. × 250 mm, 2 ml/min, n-hexane:EtOAc = 9:1) and yielded 21 (0.2 mg), 13 (0.5 mg), and a mixture (5:1) of 12 and 4 (7.9 mg). Fr. E (1.39 g) was separated by silica gel column chromatography (30 mm i.d. × 150 mm, n-hexane:EtOAc = 1:0, 98:2, 96:4, 94:6, 92:8, 9:1, 85:15, 8:2, 7:3, EtOAc, MeOH) and gave seven fractions. Fr. E-2 (637 mg) was identified as 10. Fr. E-3 (246 mg) was a mixture (14:1) of 12 and 4. Preparative HPLC of Fr. E-4 (41.8 mg) was with COSMOSIL 5SL-II (10 mm i.d. × 250 mm, 6 ml/min, n-hexane:EtOAc = 85:15) and gave 19 (0.9 mg), a mixture (2:1) of 12 and 4, and fractions E-4 to 3 and E-4 to 4. Preparative HPLC of Fr. E-4 to 3 (4.2 mg) was with YMC-Pack Diol-120-NP (4.6 mm i.d. × 250 mm, 2 ml/min, n-hexane:EtOAc = 85:15) and afforded 7 (1.0 mg). Fr. E-4 to 4 (4.5 mg) was subjected to preparative HPLC using YMC-Pack Diol-120-NP (4.6 mm i.d. × 250 mm, 2 ml/min, n-hexane:EtOAc = 85:15) and gave 4 (0.8 mg), 1 (1.3 mg), and a mixture (3:1) of 5 and 6 (1.9 mg). Preparative HPLC of Fr. E-5 (54.9 mg) and Fr. E-6 (62.9 mg) using COSMOSIL 5SL-II (10 mm i.d. × 250 mm, 6 ml/min, n-hexane:EtOAc = 85:15 for Fr. E-5; 8:2 for Fr. E-6) yielded 1 (8.5 mg) and 3 (4.6 mg) from Fr. E-5 and 18 (4.6 mg), 8 (5.4 mg), and 17 (7.3 mg) from Fr. E-6. Fractionation of Fr. H (1.10 g) via silica gel column chromatography (30 mm i.d. × 150 mm, CHCl3:MeOH = 95:5, 92:8, 9:1, 88:12, 85:15, 4:1, 7:3, 0:1) gave five subfractions. The Fr. H-3 (539 mg) was identified as 22. Further separation of Fr. H-5 (108 mg) via silica gel column chromatography (10 mm i.d. × 150 mm, CHCl3:MeOH = 4:1, 3:1, 7:3, 1:1,0:1) gave 22 (28.4 mg) and 9 (10.5 mg). Fr. H-5 to 2 (40.8 mg) was separated via preparative HPLC using COSMOSIL 5C18-PAQ (20 mm i.d. × 250 mm, 6 ml/min, H2O:MeOH = 4:6) and yielded 22 (3.3 mg) and 9 (10.0 mg).
A yellow powder, 1H and 13C NMR data: in Table 4. FT-IR cm−1: 3412, 2229, 1746, 1632, 1455, 1405, 1076. FAB-MS m/z: 427.0687 (calcd for C18H19O10S: 427.0699). MS (FAB, negative mode) m/z: 427 (M-H)−. [α]29 D + 2.3 (c = 0.9, MeOH).
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Japanese Society for the Promotion of Science (grant number 16K18897 and 20K07102).
ORCID iDs
Yoshinori Saito
Takashi Tanaka
References
1.
SchmidtGJSchillingEE. Phylogeny and biogeography of Eupatorium (asteraceae: eupatorieae) based on nuclear ITS sequence data. Am. J. Bot. 2000;87(5):716-726.
2.
ToriMTakeichiYKugaHNakashimaKSonoM. Two new chlorine-containing germacranolides, eupaglehnins E and F from Eupatorium glehni. Heterocycles. 2000;52(3):1075-1078.
3.
ToriMTakeichiYKugaHNakashimaKSonoM. Seven germacranolides, eupaglehnins A, B, C, D, E, and F, and 2α-acetoxyepitulipinolide from Eupatorium glehni. Chem. Pharm. Bull., 2002;50(9):1250-1254.
4.
ToriMMorishitaNHirotaN, et al.Sesquiterpenoids isolated from Eupatorium glehnii. Isolation of guaiaglehnin A, structure revision of hiyodorilactone B, and genetic comparison. Chem. Pharm. Bull. (Tokyo). 2008;56(5):677-681.
5.
SaitoYMukaiTIwamotoY, et al.Germacranolides and their diversity of Eupatorium heterophyllum collected in P.R. China. Chem. Pharm. Bull. (Tokyo). 2014;62(11):1092-1099.
6.
SaitoYTakiguchiKGongXKurodaCToriM. Thiophene, furans, and related aromatic compounds from Eupatorium heterophyllum. Nat. Prod. Commun. 2011;6(3):361-366.
7.
BohlmannFZderoC. Notiz über die inhaltsstoffe von Carelia cistifolia less. Chem. Ber. 1971;104(3):964-966.
8.
BohlmannFZderoCGrenzM. Natürlich vorkommende terpen-derivate, 81. Weitere inhaltsstoffe aus vertretern der Eupatorium-gruppe. Chem. Ber. 1977;110(3):1034-1041.
9.
ZderoCBohlmannFKingRM. Diterpenes and norditerpenes from the Aristeguetia group. Phytochemistry. 1991;30(9):2991-3000.
10.
AlarconSRFuenteJRDLNovaraLSosaVE. Benzofuran and coumarins in Trichocline reptans wedd. An. des la Asoc. Quimica Argentina. 1998;86(3-6):248-251.
11.
YamaguchiSNittaTHosakaTNishinoYKawaseY. The synthesis of two 6-acetyl-2,3-dihydro-5-benzofuranols naturally obtained from Baccharis conferta. Bull. Chem. Soc. Jpn. 1990;63(11):3230-3232.
12.
BohlmannFZderoC. Stevisalicinon. ein neuer diterpentyp, sowie weitere inhaltsstoffe aus Stevia-arten. Liebigs Ann. Chem. 1985(9):1764-1783.
13.
KamthongBRobertsonAFurano compoundsV. The synthesis of tetrahydroeuparin and the structure of euparin. J. Chem. Soc. 1939;925:933-936.
14.
ElsohlyMADoorenbosNJQuimbyMWKnappJESlatkinDJSchiffPL. Euparone, a new benzofuran from Ruscus aculeatus L. J. Pharm. Sci. 1974;63(10):1623-1624.
15.
GuptaSRSeshadriTRSoodGR. The structure and synthesis of neobavachalcone, a new component of Psoralea corylifolia. Phytochemistry. 1977;16(12):1995-1997.
16.
BohlmannFKleineKMBornowskiH. Polyacetylenverbindungen, XLI. Über zwei thiophenketone aus Artemisia arborescens L. Chem. Ber. 1962;95(12):2934-2938.
17.
ZalkowLHKeinanESteindelSKalyanaramanARBertrandJA. On the absolute configuration of toxol at C-3. Vicinal H-H coupling constants in 2–alkyl-3-hydroxydihydrobenzofurans. Tetrahedron Lett. 1972;13(28):2873-2876.
18.
MertesMPPowersLJShefterE. Isolation and identification of the cis–trans stereoisomers of substituted 3-hydroxy-(or acetoxy) 2-methyl-2,3-dihydrobenzofurans. Dihydrobenzofurans which obey the Karplus equation. J. Org. Chem. 1971;36(13):1805-1807.