
Abstract
With the current pandemic of the novel coronavirus disease 2019 (COVID-19) in hand, researchers around the globe are dexterously working to find the best suitable drug candidates and overcome vaccination-related challenges, to achieve efficient control over the second surge of COVID-19. The medical consultants time and again have been reiterating the need to abide by the precautionary steps to prevent the spread of the coronavirus by maintaining social distancing when outside, sanitizing hands regularly, and wearing masks and gloves. They also suggest taking a good and hygienic meal so as to boost immunity. Indians have an inborn nature of using natural spices, food, and medicines in their daily lives. Indian researchers have paid heed to deploy compounds from natural sources to explore potential antiviral agents against COVID-19 as the chances of acquiring side effects are perceived as less, and the efficacy of phytochemicals from medicinal plants is sometimes greater when compared to their synthetic counterparts. In the present study, we performed an in silico molecular docking and molecular dynamic simulation analysis of screened phytochemicals from a comprehensive list of Ayurvedic herbs/functional foods that are present in natural food products against key receptor proteins of severe acute respiratory syndrome coronavirus 2. We found that Aegle marmelos, Vetiveria zizanoides, Moringaolifera, and Punica granatum have antiviral potential to prevent coronavirus infection in the populace.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) mainly targets the human respiratory system and has created havoc all over the world. The coronavirus disease 2019 (COVID-19) is responsible for taking the lives of more than 3 million people worldwide till mid-April 2021. Despite being declared as a pandemic by WHO, there is the unavailability of effective drugs and a scarcity of vaccines to prevent viral infection, which is resurrecting its threat due to mutations in different parts of the world. Few vaccines are available in different parts of the world and others are at the trial stage. 1 However, effective prophylactic therapy is still required as vaccinating the entire population is not feasible among recurrent mutations of SARS-CoV-2. Many repurposed drugs have been hypothesized and are under clinical trials such as chloroquine, remdesivir, favipiravir, darunavir, umifenovir, nitazoxanide, and thalidomide.2,3
With the rise of computationally intelligent systems, computational biologists and researchers have deployed artificial intelligence and machine-learning algorithms to track, monitor, and diagnose COVID-19 infections around the globe. 4 Also, machine learning techniques have been used in computer-aided drug discovery and designing for COVID-19. 5 Researchers have also discerned that African extracts of Zingiberoffinale and Anacardiumoccidentale can be deployed as potential suppressants for 3C-like main proteinase (3CLpro) from SARS-CoV-2. 4 Several drugs have now been approved, such as remdesivir, dexamethasone, bamlanivimab, casirivimab, and imdevimab, 6 for curbing the viral infection. Although we now have various synthetic drugs, therapies, and vaccines to break the viral chain of COVID-19, it is still recommended to take preventive measures and may include potential antiviral herbal agents in a healthy diet. Ayurveda is a more than 3500-year-old Indian medicinal system according to which for a healthy life it is important to understand the classification of food. 7 In India, foods are commonly known as “functional foods” as the components present in them have probiotics, antioxidants, and body-healing properties. 8 The functional molecules such as vitamins, flavonoids, and carotenoids help in reducing the risk of getting the disease in humans,9,10 and thus we can say that “we are what we eat.” Many compounds from these plant products have been used in viral infection earlier.11-13
In the present study, we aimed to find the antiviral properties of Indian plant-based food against COVID-19 through in silico analysis. This makes it easier and less time-consuming in screening lead compounds and their targets. 14 We have chosen Aegle marmelos (bel/wood apple), Vetiveriazizanioides (vetiver/usheer/khus), Moringa oleifera (drum stick), and Punica granatum (pomegranate), which have antiviral properties.15-18 The selected phytochemicals were docked against 2 SARS-COV-2 target proteins: 3CLpro and RNA-dependent RNA polymerase (RdRP). These play a critical role in viral replication and transcription, making them targets for drug design and discovery.
Materials and Methods
Retrieval of Phytochemicals and Preparation of Library
To select the best of Indian foods, Ayurveda classical texts were screened thoroughly to prepare an exhaustive list of medicinal plants that are known to encapsulate strength and immunity improving properties, which may exhibit antiviral activities. Subsequently, 673 phytochemicals of these plants were retrieved from IMPPAT, 19 KNApSAcK, 20 PubChem, 21 and ChEMBL 22 databases. Compounds whose mol/.sdf were not available were drawn and converted using Online SMILES Translator and Structure File Generator (https://cactus.nci.nih.gov/translate/).
Retrieving Target Proteins
The target proteins were retrieved from RCSB PDB (https://www.rcsb.org/) with their PDB IDs including 6NUR (RdRp) and 1UJ1 (3CLpro).
Absorption, Distribution, Metabolism, Excretion, and Toxicity Analysis
In order to select the best, soluble, druggable, lead-like, and nonviolating drug compounds for the progression of this study, the drug library was subjected to clusters for ADME analysis using Swiss ADME. 23 Compounds that were qualified using ADME screening were then subjected to toxicity analysis using TEST software (https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test).
Target Optimization
Both target proteins were screened for posttranslational modification (PTM) check using the Vienna PTM tool, 24 and protein topologies were prepared using PDB2gmx of GROMACS,25,26 which were then used to select the best-optimized protein targets for molecular docking approaches.
Molecular Docking Study
Active Site Prediction
This was executed using 3DligandSite 27 and COACH-D 28 to find the active sites/binding pockets in the selected target proteins where the drugs are most likely to bind with stable free energy.
Protein Preparation
The target proteins were retrieved from PDB, and thus cannot be directly employed for molecular docking as they may be bound with heavy atoms, cocrystallized ligands, water molecules, ions, and cofactors that brings up the need for a preprocessing by adding hydrogen atoms, removing the surrounding water molecules, removing cobound ligand(s)/ions, and adding polar charges. Once the preprocessing was done, these were converted from .pdb to .pdbqt.
Preparing Compounds for Docking:
Before the phytochemicals could be deployed for molecular docking, they were energy minimized using various parameters—forcefield mmff94 optimization was employed using the steepest descent algorithm in 200 steps and the termination criteria were kept for the update if the energy difference was <0.1. Phytochemicals were converted from .mol2/.mol/.sdf to .pdbqt and were then subjected to docking in AutoDock Vina. 29 For multidrug docking, we used PyRx software tool, 30 which works for virtual screening and docking in AutoDockVina.
Docking was carried out twice, 1 for each target, with the same initial steps of preprocessing. After loading the prepared target with set parameters, the prepared ligand file was also uploaded. The grid size was set to 50 × 50 × 50 points with a grid spacing of 0.375 Å with exhaustiveness equal to 8. The Broyden–Fletcher–Goldfarb–Shanno run (BFGS run) was used as the main optimization algorithm for molecular docking.
Interaction Analysis
The complexes were further subjected to an interaction analysis using a webserver protein–ligand profiler. 31
Molecular Dynamics Simulations
The best-docked protein target and ligand complexes were subjected to refinement and molecular dynamics simulation (MDS) using CHARMM 32 and VMD, 33 respectively. The protein complexes which were .pdb complex files were converted into .psf, and trajectory files were retrieved which were then used to minimize solvate, neutralize and then refine the complex structures. Cubic water boxes with transferable intermolecular potential with 3 points water molecules were deployed for solvation of the 2 selected complexes. 34 The size of the box was selected so that there was a safe distance of 10 Å between the protein target and the corners of the periodic box. A 10 Å cutoff distance was used to compute short-range nonbonded interactions. The particle mesh Ewald method 35 was used to discern the long-range electrostatic associations. Generalized born molecular mechanics surface area was deployed to retrieve the approximate results in an explicit solvent. We have deployed NVT dynamics that hold an amount of substance (N), volume (V), and temperature (T) constants. The Noose–Hover temperature was set to 300 K and the entire simulation was executed in 1000 steps for 50 ns. Topology and force field parameters were assigned from the CHARMM27 protein–lipid parameter set for the proteins and the CHARMM General Force Field (CGenFF) parameter set for the small molecule ligand. 36
Deformability, B-Factor, and Covariance Computation
The MDS complexes were subjected to deformability, B-factor, covariance, and root-mean-square fluctuation analysis to check for any residues that may still be unstable or deformed after the coarse-grained MDSs. The deformability, B-factor, and covariance analysis were executed using iMODS software. 37
Results
Absorption, Distribution, Metabolism, Excretion, and Toxicity Analysis
The prepared drug library (n = 673) was subjected to pharmacokinetic (PK) absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis, pharmacodynamic, and physicochemical analysis. Herbal compounds that had a higher gastrointestinal absorption, solubility, blood–brain barrier permeability, good logP values (partition coefficient), and those which did not violate any of the rules namely—Lipinski's RO5, Veber's, Ghosh's, Egan's, and Mugge’s rules were considered. 23 Finally, herbal compounds that had a good bioavailability, lead-likeness score with better synthetic accessibility were selected. After ADME analysis, phytochemicals were subjected to toxicity screening to select those that exhibited no susceptible toxicity or binding to any toxic receptor.
Target Optimization
The optimization of RdRp (PDB ID: 6NUR) and 3CLpro (PDB ID: 1UJ1) was performed to prevent any steric clashes present before the protein preparation step. There were no mutated amino acids or any other PTMs present in their tertiary structure. Figure 1 depicts the tertiary structure of the 2 target proteins after optimization.
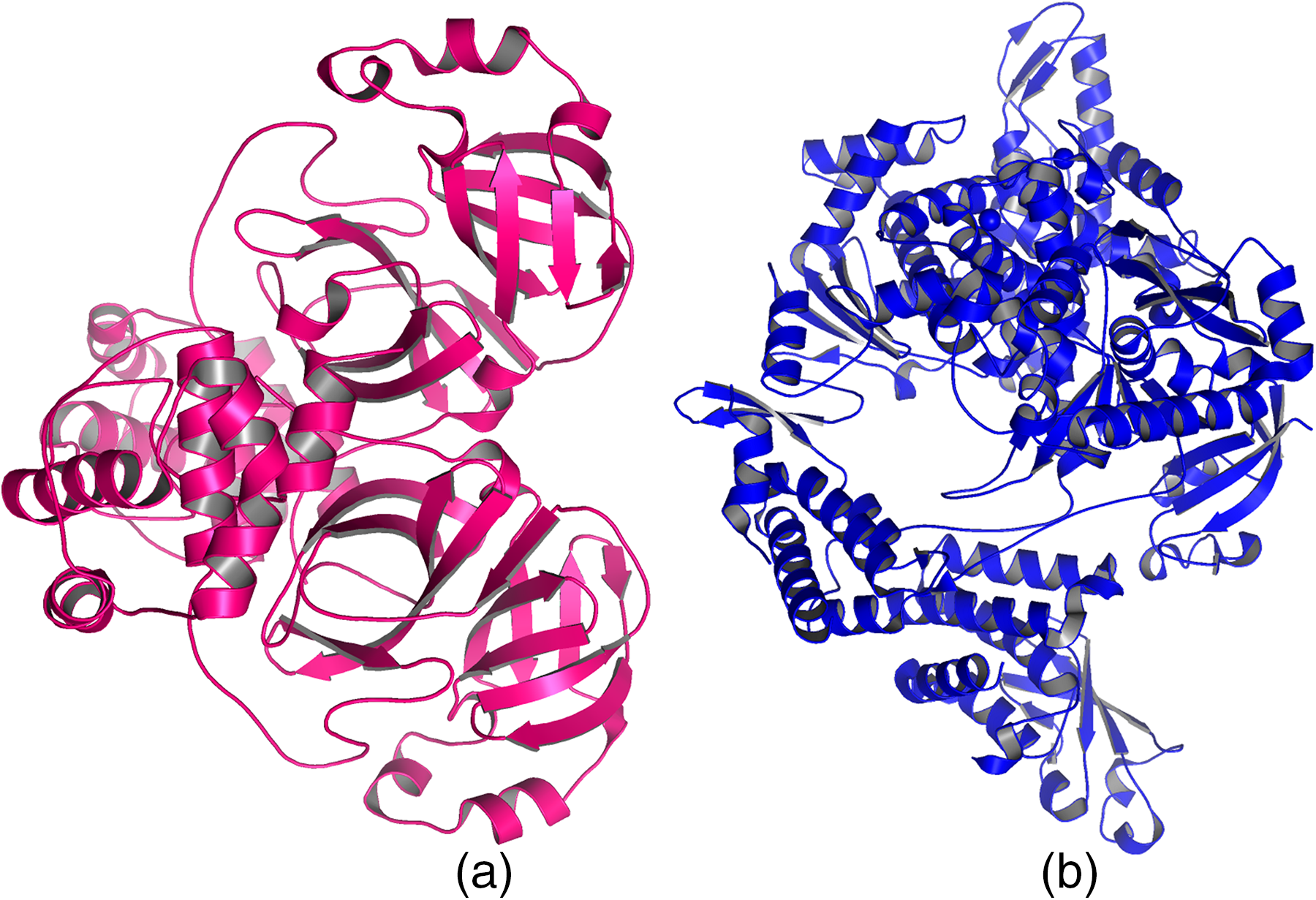
Target proteins, (a) 3CLpro (PDB ID: 1UJ1) and (b) RdRP (PDB ID: 6NUR).
Molecular Docking
In the current work, our main aim was to study natural food products that could potentially and effectively interact with the SARS-CoV-2 proteins—6NUR and1UJ1, and, therefore, could potentially be used as food alternatives for immunity boost-up against COVID-19. Out of the 673 ADMET screened compounds, only 359 passed, which were then subjected to molecular docking. Some of these phytochemicals had no .sdf/.mol structure available or were simply mentioned in the literature, or caused some parsing error while subjecting them to molecular docking analysis, and were thus dropped. Thus, out of these 359, only 100 were used for molecular docking analysis. After the docking analysis, only 4 phytochemicals out of these 100 showcased a better binding affinity towards both the target proteins—3CLpro and RdRp. These phytochemicals namely, marmin, khusinol oxide, benzylamine, and malic acid, are represented in Table 1. Figure 2 represents the docked phytochemicals to both of the target proteins—6NUR and 1UJ1. The best-docked complexes selected for MDS are those of benzylamine_3CLpro and malic acid_RdRp, respectively.
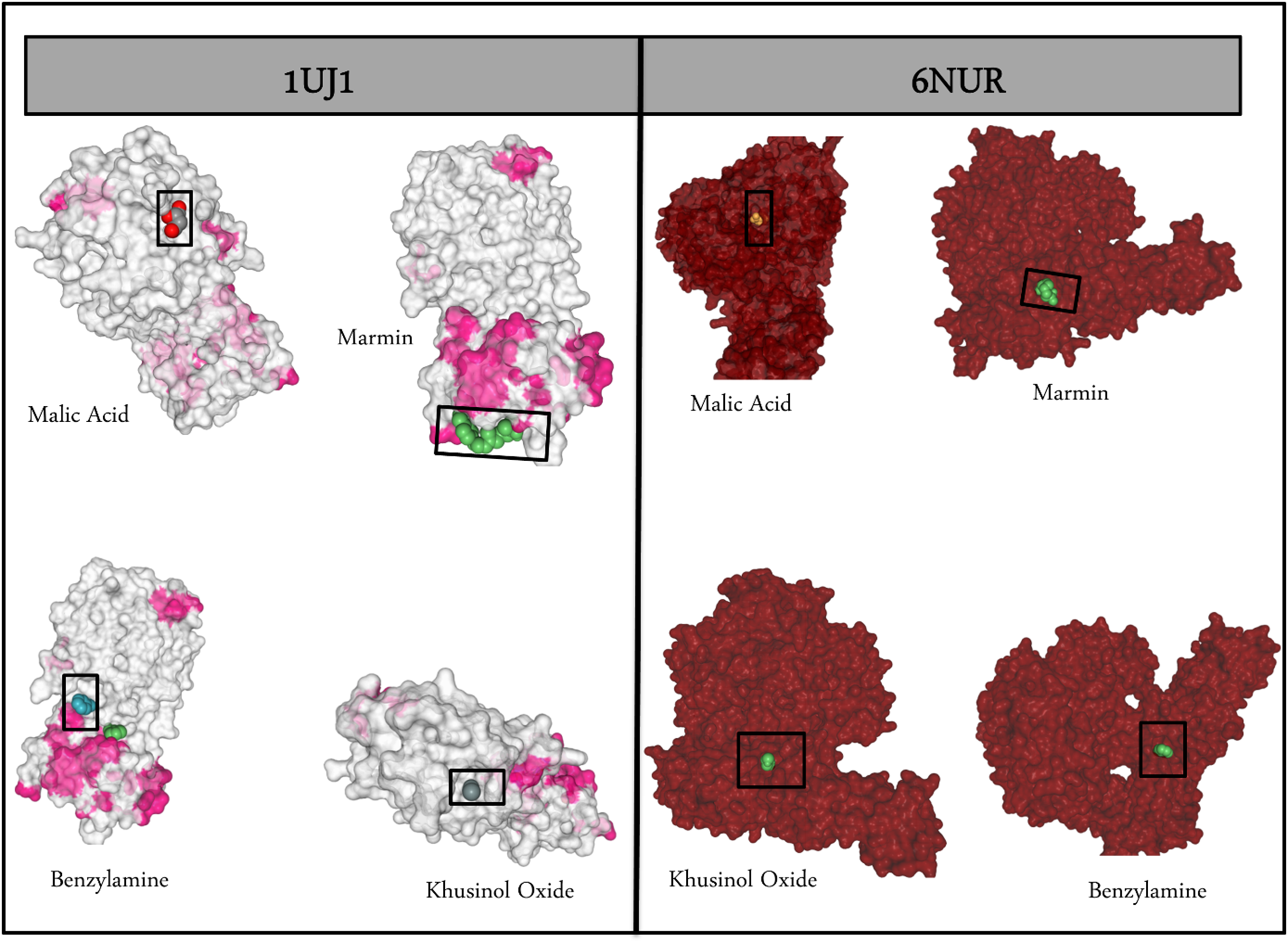
The best scoring docked complexes for target receptors 3CLpro (1UJ1) and RdRp (6NUR).
Molecular Docking Scores of the 4 Phytochemicals With the Target Proteins: 3CLpro and RdRp.
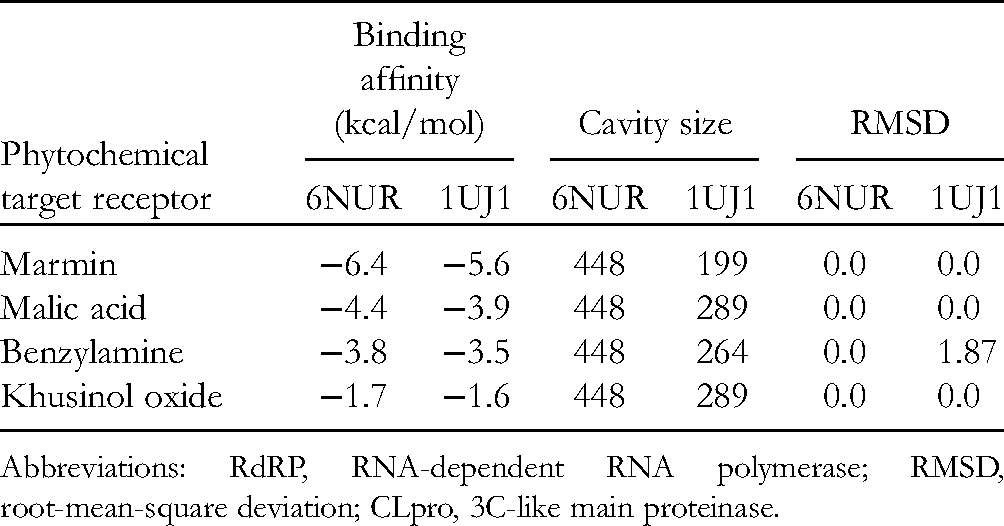
Abbreviations: RdRP, RNA-dependent RNA polymerase; RMSD, root-mean-square deviation; CLpro, 3C-like main proteinase.
Interaction Analysis
Before submitting the selected complexes for MDS at 50 ns, they were further validated using an interaction analysis to check the types and number of interactions formed between the docked phytochemicals, marmin, malic acid, benzylamine, and khusinoloxide, with the target proteins—RdRp and 3CLpro. Table 2 showcases the interaction results. A complex is considered to be strong when the number of hydrogen bonds is greater with a few hydrophobic interactions, salt bridges, and pi–pi interactions. We subjected all 4 phytochemicals docked with RdRp and 3CLpro separately to check for the number of interactions that each compound formed. The interaction analysis suggested that the complexes that had a good binding affinity were the ones that formed the highest number of hydrogen bonds.
Interaction Analysis for the 4 Phytochemicals and Target Proteins: 3CLpro and RdRp.

Abbreviations: RdRP, RNA-dependent RNA polymerase; Dist, distance; CLpro, 3C-like main proteinase; MET, methionine; ASN, asparagine; GLU, glutamic acid; GLY, glycine; TRP, tryptophan; ALA, alanine; LYS, lysine; ARG, arginine; THR, threonine; GLN, glutamine; ASP, aspartic acid; SER, serine.
Malic acid formed 3 hydrogen bonds with 3CLpro (1UJ1), 1 with glutamic acid (GLU 14A), methionine (MET 17A), and glycine (GLY 71A), while it formed 4 hydrogen bonds with RdRp (6NUR)—1 with serine (SER 15C), 2 bonds with residue glutamine (GLN 18C and 19C) and 1 with glycine (GLY 413A). A single salt bridge was also observed to be formed with residue lysine (LYS 411A). This was the only phytochemical that was associated with the formation of a greater number of hydrogen bonds with both the target proteins. Khusinol oxide was the second phytochemical that showed a variable number of hydrogen bonds. It formed 3 bonds with 3CLpro amino acid residues—arginine (ARG 188A), threonine (THR 190A), and glutamine (GLN 192A), while it was associated with only 2 residues of RdRp for hydrogen bonding, namely—serine (SER 15C) and glycine (GLY 413A). Benzylamine, on the other hand, endorsed a mix of interactions. With the 3CLpro, it formed a hydrogen bond with methionine (MET 17B), 2 hydrophobic interactions with alanine (ALA 70B) and lysine (LYS 97B), and a single pi–pi stacking interaction with tryptophan (TRP 31B). It formed a single hydrogen bond with serine (SER 15C) with RdRp. Marmin formed 2 hydrogen bonds with residues methionine (MET 17A) and asparagine (ASP 95A) and 2 hydrophobic interactions with tryptophan (TRP 31A) and alanine (ALA 70A) with 3CLpro, while a single hydrogen bond with residue threonine (THR 556A), 3 hydrophobic interactions with threonine (THR 455A), lysine (LYS 621A), aspartic acid (ASP 623A) and a single salt bridge with arginine (ARG 624A) with RdRp. It must be noticed here that malic acid and benzylamine formed a reasonable number of hydrogen bonds and significant pi–pi stacking interactions as these are known to make a protein complex stable and are relevant for molecular recognition processes. 38 The remaining phytochemicals, marmin, and khusinol oxide, formed a few hydrogen bonds, hydrophobic interactions, and salt bridges because of the conformational space occupied by them. Figure 3 depicts the interactions of these phytochemicals mentioned above with the target proteins—3CLpro and RdRp.

Interactions formed between the 4 phytochemicals and target proteins—RdRp and 3CLpro.
Molecular Dynamics Simulations
After the MDS and refinement, it is evident that the complexes have been refined to the best potential possible and the overall energy of the complex has also been stabilized with all the structures having a good root-mean-square deviation (RMSD) score. Table 3 summarizes the best-refined complexes which can be clinically evaluated further for a suitable treatment against SARS-CoV-2. From our analysis, we can discern that the best stable complex is malic acid bound to RdRp (PDB ID: 6NUR) as its overall energy is better than that of benzylamine_3CLpro. There was no “impropers” present in the complexes after refinement. The phytochemicals after refinement suggest the presence of some important residues that impact the binding of the ligand to the complexes such as tryptophan, methionine, serine, glutamine, and glycine. The superimposition and comparison of the complexes before and after refinement showcase binding mechanisms with the lowest free energy barriers in the same direction. Figure 4 represents the superimposed complexes before and after refinement.
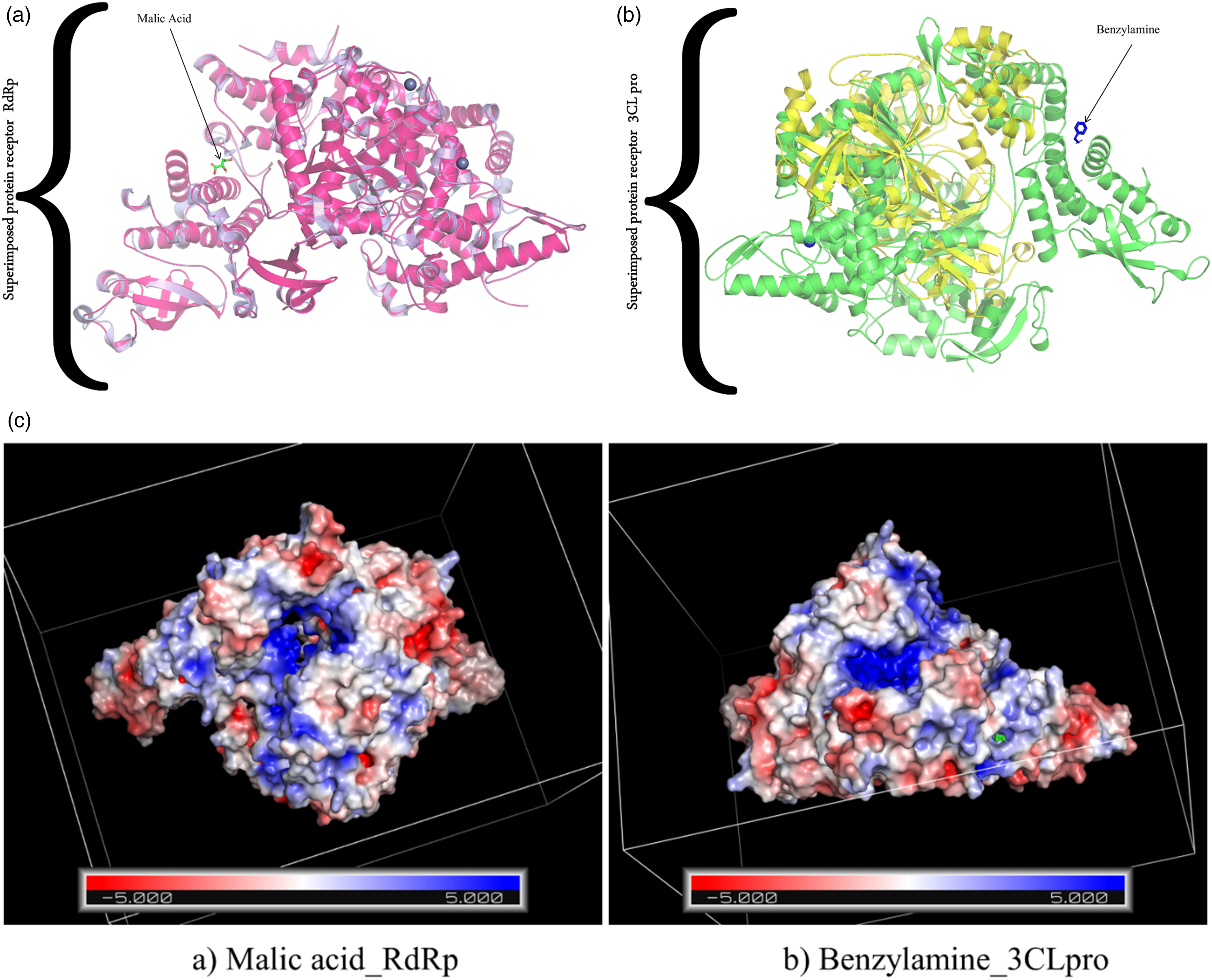
(a) The superimposition of initial structures RdRP (purple) and simulated structure (pink) with malic acid. (b) 3CLpro (yellow) and simulated structure (green color) with benzylamine. (c) MM-GBSA electrostatics is represented in the form of an APBS map.
Details of the MDS With Electrostatics Computation.

Abbreviations: MDS, molecular dynamic simulation; RMSD, root-mean-square deviation; GBSE, generalized born self energy; RdRP, RNA-dependent RNA polymerase; CLpro, 3C-like main proteinase.
The RMSD displays numerous fluctuations present in the complex during the MDS encapsulating the stability of the refined structure. It also verifies whether the simulation has been equilibrated or not. The ligand-binding pose energy and interactions formed between the protein and the phytochemical are directly related and dependent on residual fluctuation values. 39 RMSD scores were recorded to be either 0.02 for malic acid bound to RdRp and 0.31 for benzylamine with 3CLpro, indicating a few changes after the refinement of the 2 complexes. The accuracy score defines the improvement of the backbone structure of the initial protein structure. Malic acid bound to RdRp is discerned to be much similar to its original structure with few fluctuations or energy frustrations in its amino acid residues as its accuracy score is 0.988, whereas benzylamine bound to 3CLpro showed an accuracy of 0.9826. Out of the 2, malic acid–RdRp is more stable as it has fewer steric hindrances and clashes and accuracy scores (refer Table 3). The MolProbity score enables us to understand the physically correct alignment of the best-refined structure. Generally, MolProbity scores for tertiary structures fall in the range of 1 to 2 Å. Our results show that the complex malic acid–RdRp (MolProbity score = 1.623) structure has good physical correctness when compared to benzylamine-3CLpro (MolProbity score = 1.784). The electrostatic study helps in understanding how molecules associate with one another under myriad molecular environments. The Adaptive Poisson–Boltzmann Solver (APBS) software is essential in solving the equations of continuum electrostatics for large complexes, in turn aiding in understanding the chemical, biological, and biomedical applications. 40 Our study reveals that malic acid–RdRp had an APBS range between −607.381 and 512.402, while benzylamine–3CLpro recorded an APBS range between −645.067 and 516.185. The molecular mechanics generalized Born surface area continuum solvation (MM-GBSA) calculations (refer Table 3) suggest that complex malic acid–RdRp is more robust and electrostatically stable when compared to that of benzylamine–3CLpro. Figure 4(c) displays the MM-GBSA calculations in the form of an APBS map, as observed in PyMol software. 41 The radius of gyration (ROG) of a protein structure displays the root-mean-square (RMS) average of the distance of all deflecting elements from the center of mass of the structure under observation. 42 Our analysis discerns that the ROG of malic acid–RdRp is satisfactory when compared to benzylamine–3CLpro. ROG calculation was computed after the MDS phytochemical bound in the binding cavity of target proteins (refer Table 3).
Deformability, B-Factor, and Covariance Computations
Deformability is a measure of the ability of a given molecule to deform at each of its residues. This is mainly observed in the form of “highest peaks” that can be derived from high deformability regions. The major deformability was observed in the benzylamine_3CLpro complex as the complex has many peaks of approximate value 1.0 deformability index as compared to the malic acid_RdRp complex. The B-factor is calculated by taking the PDB structure and applying normal mode analysis (NMA) to it by simply multiplying the NMA mobility by 8pi2. The B-factor analysis also provides an average RMS approximate. Malic acid_RdRp is observed to be much better when compared to benzylamine_3CLpro. The covariance matrix provides how residues in the complex are correlated; the higher the correlation, the better is the complex. The red coloration indicates a good correlation between residues, white coloration depicts no correlation, while blue indicates anticorrelations. Malic acid_RdRp suggests a good correlation with a few anticorrelations in the majority, while benzylamine_3CLpro indicated more anticorrelations with a fine correlation. Figure 5 represents the above-mentioned results.

Deformability, B-factor, and covariance analysis (a) malic acid_RdRp and (b) benzylamine_3CLpro.
Discussion
The rapid spread of the COVID-19 pandemic has challenged the medical and scientific communities all over the world. Due to the emergence of new variants 43 of SARS-CoV-2, vaccination-related challenges, 44 and lack of specific preventive measures, symptomatic management is still the mainstay. In the present scenario, good nutritional food becomes important to boost the immune system against the COVID-19 virus. Green leafy vegetables increase good bacteria in the human gut which are responsible for 85% of the immune system, 45 whereas excess intake of animal food lowers the good bacteria in the gut and promotes inflammation, cardiovascular diseases, chronic obstructive pulmonary diseases, cancer, renal diseases, and diabetes. 45 Previous studies have reported that the antioxidant glutathione and a bioflavonoid, quercetin, present in plant-based food, prevent many infectious diseases. 45 Thus, identifying plant-based foods has additional benefits in terms of safety and higher translational value.
In silico studies are being employed by different researchers to screen natural botanicals based on their classical references for finding suitable lead compounds. In recent research, several attempts have been made to explore their mechanism of action and effects on the host immune system through their activity on target proteins. Molecular docking studies have validated the significance of 3CLpro and RdRp as suitable target proteins for inhibiting SARS-CoV-2 replication. 46 In the present study, 3CLpro and RdRp were targeted to find lead compounds that can act as a new drug against coronavirus. The selection of plants in the current study was based on their classical indication against fever and dosha pacifying attributes.
To find the lead molecule against SARS-CoV-2, a physicochemical library was prepared from 4 plants. These phytochemicals were subjected to molecular docking against 3CLpro and RdRp. The amino acids in 3CLpro and RdRp were exposed to various PTMs that can be either enzymatic or nonenzymatic in nature, and, thus, have the potential to completely change the original nature and functioning of the proteins, and also affect their structural dynamics and conformations. Therefore, target optimization was performed. The optimized targets were selected for molecular docking study as they had no mutations or insertions–deletions (indels), thus, would not give any misleading results. 3CLpro (PDB ID: 1UJ1) and RdRp (PDB ID: 6NUR) do not have any PTMs present in their original structure. RdRp has more helices, which suggest there are more hydrogen bonds formed within the protein chains, thus giving a polar surface to the target receptor; the main amino acids present in this target protein are—alanine, methionine, leucine, glutamate, and lysine. 3CLpro, on the other hand, has more antiparallel beta strands when compared to helices and loops, which indicate that the amino acids tyrosine, phenylalanine, tryptophan, isoleucine, valine, and threonine are present in the majority. The main binding cavity of wild type 3CLpro has amino acid residues serine (SER 215), histamine (HIS 141), and aspartic acid (ASP 163). In the mutant structure, the active residue serine (215A) is mutated to alanine (ALA) in order to prevent autocatalytic cleavage between tryptophan (TRP 264) and serine (SER 265). As far as RdRp is concerned, it is considered to be the active site base in the oxidase reaction mediated via a water molecule at position 541. Glutamic acid (GLU 361) is the base of the isomerization reaction. When mutated to glutamine, asparagine (ASN 485) is thought to be involved in positioning the water molecule.
Based on the molecular docking study, we found that, of the 4 phytochemicals, benzylamine and malic acid showcased a better binding affinity towards the 2 target receptors—3CLpro and RdRp. Previous studies have reported antipyretic and antiviral activity of various parts of A marmelos in the early stage of viral replication. 47 A 50% ethanolic extract of A marmelos fruit extract had an antiviral effect against Ranikhet disease virus, and marmalade made from the fruits has been reported to have the most effective antiviral property. 48 In the current study, marmin was found to bind to both 3CLpro and RdRpof SARS-CoV-2 virus, thus indicating A marmelos may provide immunity in the early stage of viral infection. Benzylaminephosphodiamides are being used in the prophylaxis and treatment of human immunodeficiency virus (HIV). The benzene ring has electron-donating groups which play a role in the antiviral property of benzylamine. 49 Aqueous extract of M oleifera has been reported to exhibit an inhibitory property against herpes simplex virus. 50 Since benzylamine binds to both 3CLpro and RdRp and is one of the phytochemicals from M oleifera, therefore M oleifera can be included in the regular diet. M oleifera has been used for ages in African Traditional Medicine against HIV. Malic acid is a product in Kreb's cycle and plays a role in energy generation. There is not much evidence supporting the antiviral property of malic acid, but in the present study, malic acid from P granatum (pomegranate) bound to 3CLpro and RdRp. P granatum has been reported to have antiviral properties against herpes virus, influenza virus, poxviruses, and HIV-1 virus. 51 On the basis of the present in silico results, it can be interpreted that marmin, malic acid, benzylamine, and khusinol oxide have antiviral properties making these phytochemicals good lead compounds against SARS-CoV-2. However, preclinical and clinical studies are needed to investigate the in silico predictions in biological models for further therapeutic applications.
Conclusion
In the present study, we screened 673 phytochemicals derived from 14 Indian herbs to find lead molecules for SARS-CoV-2. A total of 359 phytochemicals qualified from ADMET analysis, out of which only 4, namely marmin, khusinol oxide, benzylamine, and malic acid exhibited significant binding with RdRp and 3CLpro.These 4 phytochemicals are present in Indian foods like A marmelos, V zizanoides, M. olifera, and P granatum. Thus, the present in silico study identifies potential inhibitors of RdRp and 3CLpro of SARS-CoV-2, in Indian food, which have to be validated further, both experimentally and clinically.
Footnotes
Acknowledgments
SQ is supported by the DST-INSPIRE Fellowship provided by the Department of Science and Technology (DST), Government of India. BKK, VS, and GS would like to acknowledge the Ministry of AYUSH, Governmnet of India for their support through the Center of Excellent grant for Yoga and Ayurveda to Cener for Integrative Medicine and Research (CIMR), AIIMS, New Delhi.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.