The synthesis of a labdane oxocane epoxy-alcohol is described starting from the Wieland–Miescher ketone derivative via ring closing olefin metathesis of a diene derivative, targeting the total synthesis of a brominated oxocane labdane diterpenoid isolated from Laurencia obtusa.
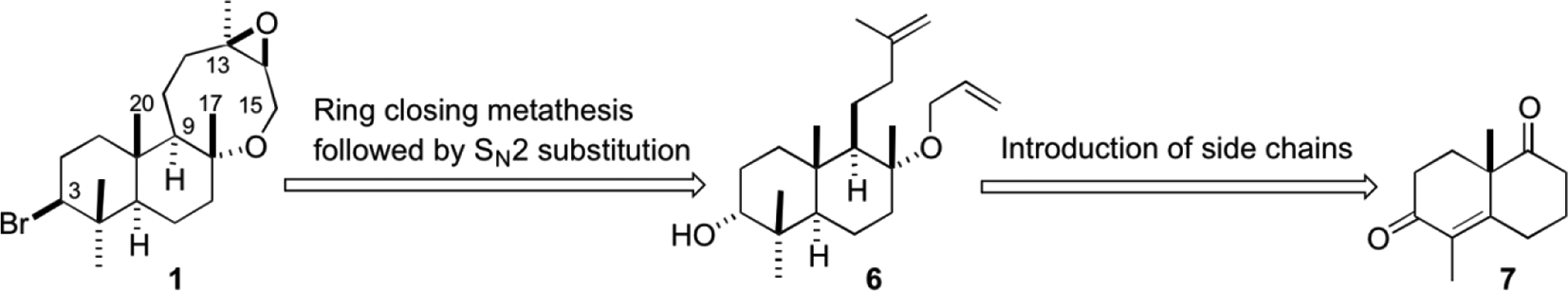
Marine organisms contain diverse secondary metabolites, some of which exhibit prominent biological activities.1,2 In particular, red algae of Laurencia species are known to produce more than 700 compounds including a variety of frameworks along with halogenated compounds.3-6 In 2003, Vagias et al isolated halogenated labdanes 1 and 2 from Laurencia obtusa obtained in the Ionian Sea of Greece,7 which possess an 8-membered or 7-membered ether ring fused with a decalin core, a labile C–Br bond at C3, and an epoxide ring or a hydroxyl group encompassing seven asymmetric centers (Figure 1). The absolute stereostructures of 1 and 2 are not yet determined.
Oxocane labdanes and congeners.
Labdane diterpenoids having oxocane or its oxidized structure are very rare; only 1 additional compound 3 has been reported from terrestrial plants.8,9 Toward a synthetic approach to these compounds, Alvarez-Manzaneda et al recently reported a transformation of (–)-sclareol (5) into compound 4 via ring closing olefin metathesis (RCM) as a key step.10
These interesting aspects, along with the unknown absolute stereostructures and the results of Alvarez-Manzaneda et al, inspired us to report our independent synthetic approach to compound 1 from Wieland–Miescher ketone derivative 7, since sclareol (5) lacks a hydroxyl group at C3. How to install the 8-membered ether ring, a labile equatorial C–Br bond at C3, and an epoxide ring at C13 with correct stereochemistries were major issues of the present synthetic project. Our retrosynthetic design is outlined in Scheme 1. Since the C–Br bond at C3 is sensitive to a variety of synthetic transformations, a route to introduce the C–Br bond at the very end of the synthetic pathway was chosen. The oxocane ring might be constructed by RCM of the diene 6, which in turn might be obtained by installation of two olefinic side chains into the Wieland–Miescher ketone derivative 7.11
Retrosynthetic design.
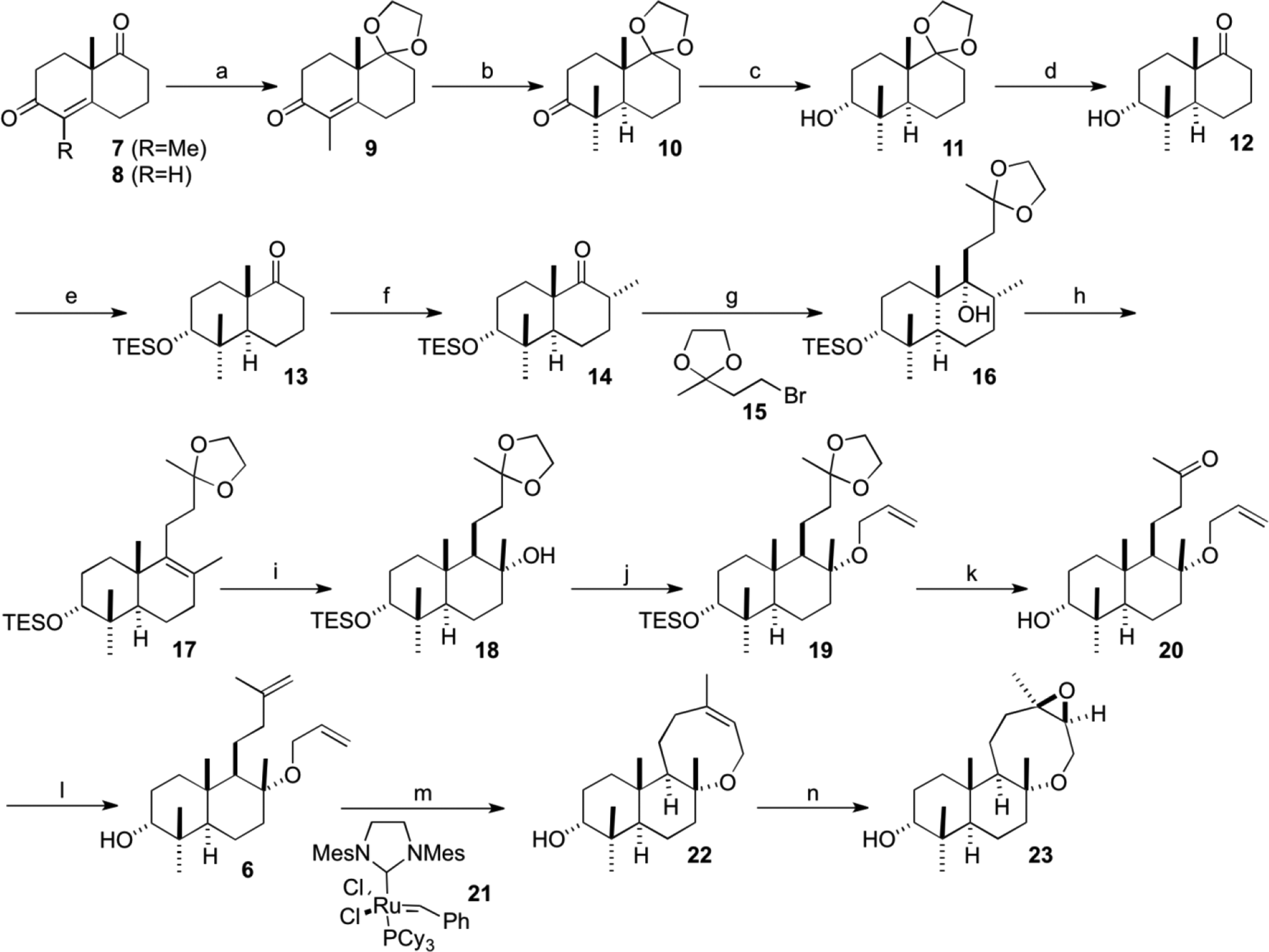
Although the hydroxyketone 12 is a known compound derived from Wieland–Miescher ketone (8) and was employed as a starting material for synthetic studies of several natural products,12-14 we took an alternative Wieland–Miescher ketone derivative 7 as our starting material, which is easy to obtain in high enantiomeric excess (Scheme 2).11
Regioselective protection of the carbonyl group at C9 of the ketone 7 was carried out simply with ethylene glycol in the presence of a catalytic amount of d-camphorsulfonic acid without employing solvent under reduced pressure at 40°C. The protocol is more favorable in terms of atom economy and environmental friendliness than the previous procedure employing 2-ethyl-2-methyl-1,3-dioxolane.11 Reductive methylation with lithium in liquid ammonia was carried out by the conventional method11 to give the trans-decalone 10 in 87% yield. In order to introduce the equatorial C–Br bond at C3 by SN2 substitution at the end of the synthetic sequence, the introduction of axial alcohol was required at this stage. Because the reduction of the carbonyl group at C3 of the decalone 10 with sodium borohydride or lithium aluminium hydride was known to give an equatorial hydroxyl group mainly,11 reduction with l-selectride was chosen to afford the α-hydroxyl compound 11 with perfect stereoselection in quantitative yield. Axial orientation of the hydroxyl group was apparent from a narrow coupling pattern of the proton at C3. Reduction with the more bulky LS-selectride resulted mainly in recovery of the starting ketone 10. Deprotection of the ketal provided keto-alcohol 12 in quantitative yield. Protection of the hydroxyl group at C3 for subsequent transformations was troublesome due to sterically encumbered axial orientation and neopentyl position, which was solved with triethylsilyl chloride in the presence of imidazole in dimethylformamide to afford triethylsilyl (TES) ether 13. An attempt to protect with triisopropylsilyl chloride resulted in the formation of a large amount of silylenol ether at C9. Methylation at C8 and subsequent equilibration to equatorial orientation gave rise to ketone 14. The Grignard reagent derived15 from bromide 15 added from the β-face of the ketone 14 selectively to give alcohol 16 in 93% yield. Purification of the bromide 15 prior to the Grignard reaction was key to the success of this reaction. Dehydration of the alcohol 16 with thionyl chloride proceeded without any trouble to give olefin 17 in 93% yield. Attempts to prepare the olefin 17 via Mizoroki–Heck reaction of the enol triflate of the ketone 14 with 3-buten-2-one derivatives all failed. Hydroboration of the double bond with BH3⋅THF at C8 of the olefin 17 proceeded mainly from the α-face to afford 8α-alcohol 18 in 56% yield. The stereochemistry of the α-hydroxyl group was determined by the nuclear overhauser effect (NOE) correlation between the C17 and C20 methyl groups. Hydroboration with 9-BBN or BH3⋅Me2S recovered the starting olefin 17. Treatment of the alcohol 18 with allyl bromide in the presence of potassium hydride, tetrabutylammonium iodide and hexamethylphosphoramide provided the O-allyl ether 19 in 70% yield. Subsequent deprotection of the ketal and TES ether afforded hydroxy-ketone 20. Wittig methylenation then gave rise the bis-olefin 6 in 91% yield.
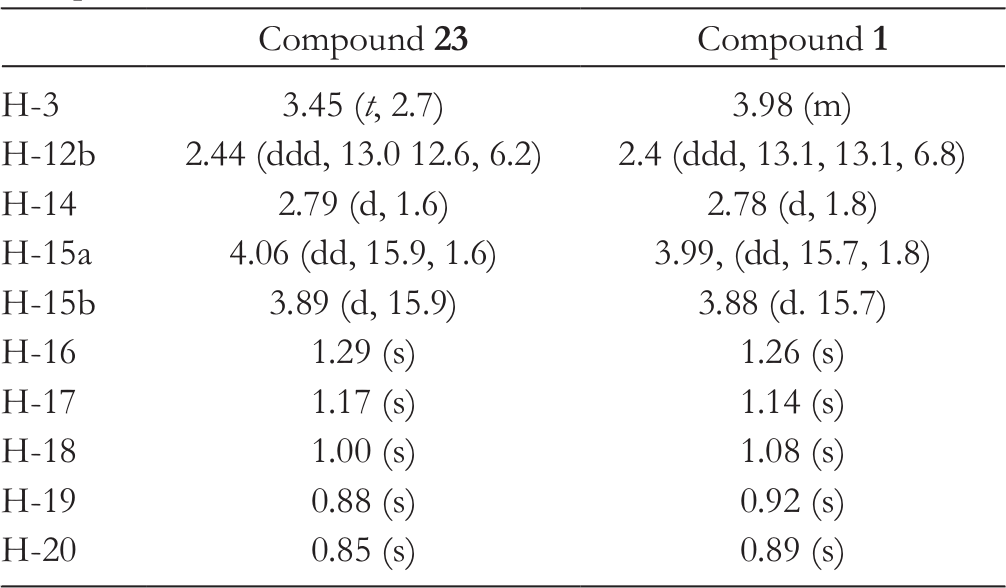
With the key bis-olefin 6 for RCM in hand, the reaction was carried out successfully employing Grubbs second-generation catalyst 21 at 40°C to afford 8-membered cyclic ether 22 in 96% yield, while the allyl group of the bis-olefin 6 was lost to some extent under refluxing conditions. Grubbs first-generation catalyst recovered the starting bis-olefin 6. Epoxidation proceeded from the β-face of oxocane 22 to provide epoxide 23, in which the stereochemistry was determined by NOE correlation between the protons at C14 and C15α (Figure 2). Similarity of 1H-nuclear magnetic resonance data of the hydroxy-epoxide 23 and the natural product 1 supported that the relative stereochemistries of both compounds were almost the same except at C3 (Table 1). Stable conformation of the oxocane 22 was anticipated to be ladder like according to basic MM2 calculations, in which β-face of the olefin of the oxocane 22 was open.
Major nuclear overhauser effect correlations.
1H-Nuclear Magnetic Resonance Data of Synthetic Compound 23 and Natural Product 1.
Compound 23
Compound 1
H-3
3.45 (t, 2.7)
3.98 (m)
H-12b
2.44 (ddd, 13.0 12.6, 6.2)
2.4 (ddd, 13.1, 13.1, 6.8)
H-14
2.79 (d, 1.6)
2.78 (d, 1.8)
H-15a
4.06 (dd, 15.9, 1.6)
3.99, (dd, 15.7, 1.8)
H-15b
3.89 (d, 15.9)
3.88 (d. 15.7)
H-16
1.29 (s)
1.26 (s)
H-17
1.17 (s)
1.14 (s)
H-18
1.00 (s)
1.08 (s)
H-19
0.88 (s)
0.92 (s)
H-20
0.85 (s)
0.89 (s)
Unfortunately, despite much effort, all our attempts at SN2 bromination of the hydroxyl group at C3 of 23 with thionyl bromide (SOBr2) or trioctylphosphine/tetrabromomethane (CBr4) resulted mainly in the migration of the methyl group at C3, ring contraction of the 6-membered ring, or decomposition. Model experiments employing alcohol 11 with phosphorus tribromide, thionyl chloride/bromine/1,2-bis(diphenylphosphino)ethane, triphenylphosphine/CBr4, SOBr2, methanesulfonyl chloride, or triflic anhydride all failed.
In summary, targeting the total synthesis of the brominated labdane 1 isolated from Laurencia obtusa, the synthesis of the labdane oxocane alcohol 23 was accomplished in 11 steps in overall 14% yield via RCM of the bis-olefin 6, which was obtained from the known hydroxyketone 12 derived from the Wieland–Miescher ketone derivative 7. The final intended bromo-dehydroxylation of the alcohol 23 was unsuccessful (supplementary material 1).
Supplemental Material
Supplementary Material 1 - Supplemental material for A Synthetic Approach Toward a Brominated Oxocane Labdane Diterpenoid Isolated From Laurencia obtusa
Supplemental material, Supplementary Material 1, for A Synthetic Approach Toward a Brominated Oxocane Labdane Diterpenoid Isolated From Laurencia obtusa by Hisahiro Hagiwara, Shohei Fujiwara, Chikako Iibachi, Toshio Suzuki and Takashi Hoshi in Natural Product Communications
Footnotes
Author note
This paper is dedicated to the late Professor Kenji Mori, to whom we owe gratitude for inspiring and memorable encouragement.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
HarizaniM.IoannouE.RoussisV. The Laurencia paradox: an endless source of chemodiversity. Prog Chem Org Nat Prod. 2016;102:91-252.doi:10.1007/978-3-319-33172-0_2
4.
JiN-Y.WangB-G. Nonhalogenated organic molecules from Laurencia algae. Phytochem Rev. 2014;13(3):653-670.doi:10.1007/s11101-013-9326-0
5.
CabritaMT.ValeC.RauterAP. Halogenated compounds from marine algae. Mar Drugs. 2010;8(8):2301-2317.doi:10.3390/md8082301
6.
GribbleGW. Biological activity of recently discovered halogenated marine natural products. Mar Drugs. 2015;13(7):4044-4136.doi:10.3390/md13074044
7.
IliopoulouD.MihopoulosN.RoussisV.VagiasC. New brominated labdane diterpenes from the red alga Laurencia obtusa. J Nat Prod. 2003;66(9):1225-1228.doi:10.1021/np0301184
8.
SebastiãoNN.CordeiroIJS.dos SantosAFet al. 8,15-epoxylabdane and norlabdane diterpenoids from Eragrostis viscosa. Phytochemistry. 2010;71(7):798-803.doi:10.1016/j.phytochem.2010.02.003
9.
SebastiãoNN..FernandesN.VieiraLet al. Three new labdanes isolated from Eragrostis viscosa. J Braz Chem Soc. 2012;23(10):1940-1950.doi:10.1590/S0103-50532012005000067
10.
TorresA.GutierrezP.Alvarez-ManzanedaR.ChahbounR.Alvarez-ManzanedaE. Preparation of oxocene terpenes. The first enantiospecific synthesis of cytotoxic arenaran A. Org Biomol Chem. 2016;14(41):9836-9845.doi:10.1039/C6OB01640E
11.
HagiwaraH.UdaH. Optically pure (4aS)-(+)- or (4aR)-(-)-1,4a-dimethyl-4,4a,7,8-tetrahydronaphthalene-2,5(3H,6H)-dione and its use in the synthesis of an inhibitor of steroid biosynthesis. J Org Chem. 1988;53(10):2308-2311.doi:10.1021/jo00245a033
12.
KendeAS.DengW-P.ZhongM.GuoX-C. Enantioselective total synthesis and structure revision of spirodihydrobenzofuranlactam 1. total synthesis of stachybotrylactam. Org Lett. 2003;5(10):1785-1788.doi:10.1021/ol030039j
13.
DetheDH.DherangeBD.AliS.ParsutkarMM. Enantiospecific total syntheses of meroterpenoids (−)-F1839-I and (−)-corallidictyals B and D. Org Biomol Chem. 2017;15(1):65-68.doi:10.1039/C6OB02322C
14.
HeC.HuJ.WuY.DingH. Total Syntheses of Highly Oxidized ent-Kaurenoids Pharicin A, Pharicinin B, 7-O-Acetylpseurata C, and Pseurata C: A [5+2] Cascade Approach. J Am Chem Soc. 2017;139(17):6098-6101.doi:10.1021/jacs.7b02746
15.
PetroskiRJ. Efficient preparation of 2-methyl-1,3-dioxolane-2-ethanol and 2-(2-bromoethyl)-2- methyl-1,3-dioxolane from 4-hydroxy-2-butanone. Synth Commun. 2002;32(3):449-455.doi:10.1081/SCC-120002130
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.