Formation of a Sterically Crowded 1,6,6αλ 4 -Triselenapentalene and 4 H -Selenopyran-4-selones Fused with Two Bornane Skeletons Through the Reaction of d -Camphor p -Toluenesulfonylhydrazone With a Base and Elemental Selenium
Open accessResearch articleFirst published online February, 2020
Formation of a Sterically Crowded 1,6,6αλ 4 -Triselenapentalene and 4 H -Selenopyran-4-selones Fused with Two Bornane Skeletons Through the Reaction of d -Camphor p -Toluenesulfonylhydrazone With a Base and Elemental Selenium
Reaction of d-camphor p-toluenesulfonylhydrazone with t-butoxide and elemental selenium in dimethylformamide at an elevated temperature afforded a stable compound having a unique 1,6,6αλ4-triselenapentalene ring and 4H-selenopyran-4-selones along with dialkenyl diselenide, dibornylenes, and 1,2,5-triselenepin, and the structural confirmation of these products were carried out by X-ray crystallographic analysis. The sterically crowded 1,6,6aλ4-triselenapentalene ring fused with two bornane sleketons was stable enough under aerobic exposure and was inactive toward sodium borohydride reduction but was converted into 1,2-diselenole derivative through m-chloroperbenzoic acid oxidation.
Dialkenyl dichalcogenides have been widely recognized as the versatile synthetic precursors of various chalcogen-containing heterocycles and chalcogenocarbonyl compounds,1-9 and the recent synthetic interests have been concentrated to their higher-row derivatives. However, in spite of the potentiality of alkeneselenolate and alkenetellurolate ions as the synthetic equivalents of selenocarbonyl and tellurocarbonyl compounds,10-17 only limited studies on the generation of these alkenechalcogenolate ions have been carried out previously and the lack of general and convenient synthetic methods of these compounds has restrained the extension on the novel conversion of these compounds into chalcogen-containing heterocyclic compounds. In the course of our successive studies on the synthesis of higher-row chalcogenocarbonyl compounds, we have found a convenient synthesis of dialkenyl diselenides C through the reaction of p-toluenesulfonylhydrazones A, derived from ketones possessing an α-methylene group, with a base and elemental selenium.18-24 The reaction was assumed to proceed through the in situ generated alkeneselenolate ions Bvia Bamford–Stevens type reaction of p-toluenesulfonylhydrazones25-33 as shown in Scheme 1, and, therefore, the synthetic application of this sequence to the preparation of various selenium-containing heterocyclic compounds was keenly expected.
Synthesis of dialkenyl diselenides C via the reaction of ketone p-toluenesulfonylhydrazone with a base and elemental selenium.
Especially, our interests have been concentrated to the application of the reaction sequence by using the bornane derivatives because d-camphor and its functionalized derivatives have been widely recognized as the versatile substrates for chiral auxiliaries, chiral ligands, and chiral building blocks for asymmetric syntheses due to their commercial availability with the structural stability and its chirality.34-37 However, in spite of the extensive research works on the functionalization of bornane skeletons within these decades, there found only limited studies on the selenium-functionalized bornane skeletons except for our previous works on the preparation and the conversion of substituted bornane-2-selones. Our previous attempts for the synthesis of dibornenyl diselenide using our procedure starting from d-camphor p-toluenesulfonylhydrazone (1) also resulted in the complex mixture containing dibornylene (3), 1,4-diselenin (4), 1,2,5-triselenepin (5), and a few uncharacterized compounds along with the formation of dibornenyl diselenide (2).18 Therefore, we just started the investigation of the structural confirmation of these uncharacterized byproducts in order to obtain a key for the optimization of the reaction conditions, and after several efforts, we could isolate and characterize the structures of some of these byproducts involving a hitherto unknown purple-colored stable crystalline compounds, 1,6,6αλ4-triselenapentalene (6), and an isomeric mixture of greenish brown-colored 4H-selenopyran-4-selones (7, 8) fused with two bornane skeletons.
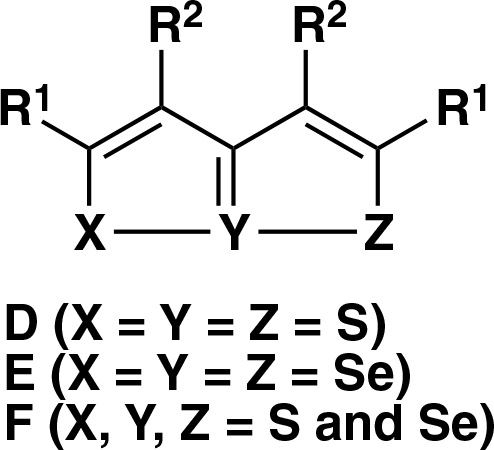
1,6,6αλ4-Triselenapentalenes D and their oxygen or nitrogen analogs are classified to be bicyclic 10-S-3 type sulfuranes having three sulfur atoms with a central hypervalent sulfur atom in the heteroaromatic ring system, and their structures, chemical reactivity, and their synthetic methods have been extensively investigated during these decades.38-50 However, in contrast to 1,6,6αλ4-trithiapentalenes D, only a few synthetic studies on their selenium analogs, such as 1,6,6αλ4-triselenapentalenes E,51-55 1,6,6αλ4-thiadiselenapentalenes, and 1,6,6αλ4-dithiaselenapentalenes F,56-58 have been carried out to date may be due to the difficulty in the preparation of suitable 1,3,5-triselenoxo intermediates. The general structures of compounds D-F are shown in Figure 1. In this report, we would like to describe the isolation and characterization of new 1,6,6αλ4-triselenapentalene (6) and 4H-selenopyran-4-selones (7, 8) fused with two bornane skeletons as the minor products of the reaction of d-camphor p-toluenesulfonylhydrazone (1) with t-butoxide (t-BuOK) and elemental selenium at an elevated temperature. A novel oxidative selenium-oxygen replacement of 6 forming 1,2-diselenole (11) bearing a neighboring carbonyl group is also reported in this paper.
Structures of 1,6,6αλ4-trichalcogenapentalenes D, E, and F.
d-Camphor was at first converted into the corresponding p-toluenesulfonylhydrazones (1) according to the usual procedure. Subsequently, a hexamethylphosphoric triamide (HMPA) or a dimethylformamide (DMF) solution of d-camphor p-toluenesulfonylhydrazone (1) was treated with t-BuOK (2.5 mol amt.) and elemental sulfur or selenium (2.5 mol amt.) at 110°C-140°C for 3-24 hours followed by an aerobic exposure of the resulting reaction mixture at room temperature,18 and only in the cases using elemental selenium, small amounts of several selenium-containing products were isolated besides dialkenyl diselenide (2), an E,Z-mixture of dimeric olefins (3),59,60 and a few uncharacterized products. On the other hand, the same reactions performed at a higher temperature over 160°C only afforded a complex mixture, and the similar reaction using elemental sulfur only afforded a complex mixture.
The mixture of less polar selenium-containing products obtained as orange oil gradually underwent decomposition with selenium extrusion during the workup procedure to form a separable mixture of relatively stable two compounds, pale yellow solids, and orange solids. All physical and spectral data for the former and the latter compound, involving mass spectrometry (MS), infrared (IR), 1H nuclear magnetic resonance (NMR), 13C NMR, and elemental analysis, were fully consistent with 1,2- or 1,4-diselenin (4) and symmetrical 1,2,5-triselenepin (5),18 respectively. Especially, the 1H NMR spectra of both 4 and 5 reveal similar symmetrical patterns with only 3 singlet signals assignable to the methyl groups of the bornane skeletons, ie, 0.80 ppm (6 H, s), 0.81 ppm (6 H, s), and 1.00 ppm (6 H, s) for compound 4, and 0.82 ppm (6 H, s), 0.87 ppm (6 H, s), and 1.05 ppm (6 H, s) for compound 5, despite the difference in their parent ion peaks in the mass spectra, ie, m/z 428 (M+) for 4 and m/z 507 (M+) for 5, respectively. Furthermore, physical and spectral data of compound 5 were identical with those of 1,2,5-triselenepin 5 reported previously by us.18
The other three compounds were also subjected to chromatographic separation, and all physical and spectral data for these compounds involving MS, IR, 1H NMR, 13C NMR, and elemental analysis were also fully assignable to symmetrical 1,6,6αλ4-triselenapentalene (6) (purple crystals), symmetrical 4H-selenopyran-4-selone (7) (green crystals), and unsymmetrical 4H-selenopyran-4-selone (8) (green crystals). The 1H NMR spectra of both 6 and 7 revealed similar symmetrical spectral patterns with only 3 methyl signals of two bornane skeletons, ie 0.68 ppm (6 H, s), 0.99 ppm (6 H, s), and 1.32 ppm (6 H, s) for 6, and 0.75 ppm (6 H, s), 0.96 ppm (6 H, s), and 1.28 ppm (6 H, s) for 7. Especially, the 77Se NMR spectrum of 6 revealed a characteristic symmetrical Se-Se-Se patterns of 572.3 ppm (d, JSe-Se = 294 Hz) and 1079.5 ppm (t, JSe-Se = 294 Hz) attributed to the 1,6,6αλ4-triselenapentalene core. On the other hand, the 1H NMR spectral pattern of compound 8 revealed an unsymmetrical spectral pattern with 6 methyl signals, ie, 0.74 ppm (3 H, s), 0.75 ppm (3 H, s), 0.94 ppm (3 H, s), 0.95 ppm (3 H, s), 1.26 ppm (3 H, s), and 1.74 ppm (3 H, s). It is worth noting that both 7 and 8 are green crystalline compounds with weak absorption peaks assignable to the n–π* transition of the selenocarbonyl functionality at 619 nm (ε = 370) for 7 and 723 nm (ε = 380) for 8, respectively, in the visible light region.
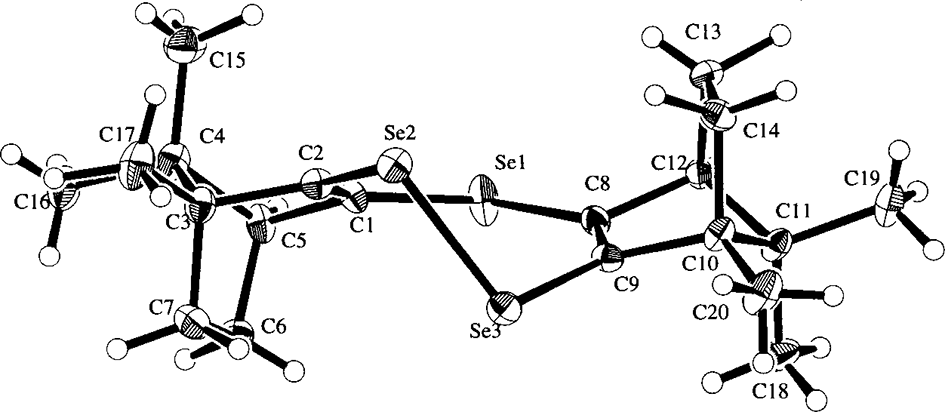
The structures of these compounds were finally determined by X-ray crystallographic analyses to be symmetrical 1,2,5-triselenepin 5, 1,6,6αλ4-triselenapentalene 6, and unsymmetrical 4H-selenopyran-4-selone 8, and their Oak Ridge Thermal-Ellipsoid Plot Program (ORTEP) drawings of 5, 6, and 8 are depicted in Figures 2–4, respectively. On the other hand, the X-ray analysis of 4 and 7 was not successful. However, 1,2,5-triselenepin 5 was gradually converted into compound 4 along with selenium extrusion by doping 5 in silica gel at room temperature for a long time as shown in Scheme 2, and these results strongly suggest the structure of compound 4 to be symmetrical 1,4-diselenin. Therefore, the alternative possible structures, such as 1,2-diselenin 4′61,62 formed through [3,3] sigmatropic rearrangement of dialkenyl diselenide 2 and the isomeric unsymmetrical 1,4-diselenin 4″, are excluded.
An ORTEP drawing of 1,2,5-triselenepin 5.
Conversion of symmetrical 1,2,5-triselenepin (5) into 1,4-diselenin (4).
An ORTEP drawing of 1,6,6αλ4-triselenapentalene (6).
An ORTEP drawing of 4H-selenopyran-4-selone (8).
According to the result of X-ray crystal data of 6, the 1,6,6αλ4-triselenapentalene ring system of 6 possesses a planar and symmetrical structure with a linear arrangement of Se(1)-Se(2)-Se(3) atoms and almost parallel 3 carbon–selenium bonds with the right Se-Se-C bond angles, ie, 88.2° for Se(1)-Se(2)-C(11), 88.8° for Se(3)-Se(2)-C(11), 87.0° for Se(2)-Se(3)-C(2), and 87.2° for Se(2)-Se(1)-C(13), respectively. Furthermore, the bond lengths of Se(1)-Se(2) and Se(2)-Se(3) were equal and the bond lengths of the central hypervalent Se(2) and the C(11) atom were also similar to those of Se(1)-C(13) and Se(3)-C(2) bonds, ie 1.82 Å for Se(1)-C(13), 1.92 Å for Se(2)-C(11), and 1.84 Å for Se(3)-C(2), respectively. In addition, the bond lengths of C(2)-C(1), C(1)-C(11), C(11)-C(12), and C(12)-C(13) were almost equal each other. These X-ray data indicated that the 1,6,6αλ4-triselenapentalene ring system possesses the heteroaromaticity. Therefore, the high field shift of 1 singlet signal of the methyl group (δ = 0.68 ppm) in the 1H NMR spectrum of 6 was explained by the anisotropic effect of the 1,6,6αλ4-triselenapentalene ring, and the existence of the 3 sp2 carbon signals (δ = 152.9, 169.2, and 195.2 ppm) in the 13C NMR spectrum of 6 was also explained by the central sp2 carbon atom (=C(11)) of the heterocyclic ring of 6. The 77Se NMR spectrum of 6 also revealed 2 selenium signals (δ = 573.2 and 1079.5 ppm) with a symmetrical Se-Se-Se coupling pattern, and the evidences for bond switching between the Se(1)-Se(2) and the Se(2)-Se(3) bonds of 6 through a tautomeric equilibration between 6′ and 6″ as shown in Scheme 3 were not observed from these NMR spectra. All the results suggest the heteroaromatic stabilization of 1,6,6αλ4-triselenapentalene ring of 6.
Possible bond-switching tautomerization for compound 6.
In contrast, the heterocyclic ring system of 8 was confirmed to be an unexpected 4H-selenopyran-4-selone ring with unsymmeterically fused two bornane skeletons. Especially, the bond lengths of C(1)=Se(1) were almost equal to those of C(3)-Se(2) and C(4)-Se(2) of the core heterocyclic ring system, ie, 1.839 Å for C(1)=Se(1), 1.846 Å for C(3)-Se(2), and 1.81 Å for C(4)-Se(2), respectively, and the bond lengths of the carbon–carbon bonds of the ring system were also much similar to each other, ie, 1.39 Å for C(1)-C(2), 1.42 Å for C(1)-C(5), 1.37 Å for C(2)=C(3), and 1.39 Å for C(4)=C(5), respectively. In addition, the 4H-selenopyran ring was almost planar with small torsion angles of the 4 atoms of the ring, ie, −1° for Se(2)-C(3)-C(2)-C(1), 7° for C(2)-C(3)-Se(2)-C(4), −1° for C(3)-Se(2)-C(4)-C(5), and 11° for Se(2)-C(4)-C(5)-C(1), respectively. These results strongly suggested that the 4H-selenopyran-4-selone ring of 8 has a contribution of the tautomeric structure of heteroaromatic selenopyrylium ion 8′ as shown in Scheme 4.
Structures of 4H-selenopyran-4-selone (8) and its tautomeric selenopyrylium ion (8′).
Selones 7 and 8 were unstable toward exposure to air and underwent gradual decomposition to afford the corresponding ketones quantitatively along with extrusion of elemental selenium. All physical and spectral data for these products involving MS, IR, 1H NMR, 13C NMR, and elemental analysis were fully consistent to symmetrical and unsymmetrical 4H-selenopyran-4-ones 9 and 10, respectively.63,64 In contrast, compound 6 was stable enough toward the exposure to air, sunlight, heating, and sodium borohydride (NaBH4) reduction. The stability of 6 toward such reagents might also be explained by the heteroaromatic character of the 1,6,6aλ4-triselenapentalene ring system. However, when a CHCl3 solution of 6 was treated with m-chloroperbenzoic acid (mCPBA, 3.0 mol amt.) at room temperature for 0.5 hours, 1,2-diselenole (11) was obtained in 81% yield along with 4H-selenopyran-4-one (9) (trace) and some uncharacterized products (trace) along with the recovery of 6 (6%) as shown in Scheme 5. Therefore, the structure of selone 7 was confirmed to be a 4H-selenopyran-4-selone with symmetrically fused two bornane skeletons. The mass spectrum of 11 revealed the parent ion peak at m/e 456 as the base peak, and the IR spectrum of 11 reveals a strong C = O streching band at 1714 cm−1. The carbonyl carbon signal was also observed at δ = 204.5 ppm in its 13C NMR spectrum, and all other physical and spectral data involving the 1H NMR, 13C NMR, and elemental analysis data were fully consistent to the structure of 11 bearing a carbonyl group at the C-2 position of one bornane skeleton. Especially, the 1H NMR and 13C NMR spectra of 11 revealed the unsymmeterical patterns, and the structure of symmetrical cyclic diselenide 11′ was excluded. It was assumed that the unusual selenium-oxygen replacement of 1,6,6aλ4-triselenapentalene ring forming 1,2-diselenole ring and a carbonyl group would proceed through oxidation of an electron-rich selenium atom, Se(1) or Se(3) of 6, via the plausible bond-switching tautomerization between 6′ and 6″.38-50 In addition, the structure of 11, bearing an isolated carbonyl group, strongly suggested that the formation of 1,6,6αλ4-oxadiselenapentalene ring would be unfavorable due to the weak attractive interaction between the selenium atoms of the 1,2-diselenole ring and the oxygen atom of the neighboring carbonyl group.
Oxidation of 1,6,6aλ4-triselenapentalene (6) and 4H-selenopyran-4-selones (7, 8).
In contrast to the case of d-campor p-toluenesulfonylhydrazone (1), similar treatment of cyclopentanone p-toluenesulfonylhydrazone with t-BuOK and elemental selenium at 110°C for 3 hours only afforded the corresponding dialkenyl diselenide in 35% yield along with the formation of a complex mixture of uncharacterized products, and neither 1,6,6αλ4-triselenapentalene nor 4H-selenopyen-4-selones was found at all in the mixture. This result strongly suggested that the steric protection by the bulky bornane substituents was much effective for the stabilization of selenium-containing products 5-8. However, attempts for trapping of the precursors of 6-8 by applying various trapping agents were not successful at all even in the case starting from d-camphor p-toluenesulfonylhydrazone (1).
The reaction pathway for the formation of products 4-8 remains unclear. However, dibornenyl diselenide 2 was mainly obtained and 4-8 were not found at all in the crude product when the reaction of 1 with t-BuOK and elemental selenium at 140°C for 3 hours, and the prolong reaction time for the same reaction, in turn, afforded 4-8 along with lowering of the yield of 2 as presented in Table 1. These experimental results suggested that 4-8 are just the secondary products formed from borneneselenolate ion B and the alternative pathway via 1,2,3-selenadiazole and/or selenirene intermediates from 1 would be negligible. Therefore, it is assumed that the products 4 and 5 would be afforded through a plausible pathway involving the in situ generation of β-selenoxoselenolate ion G formed through the reaction of borneneselenolate ion B with elemental selenium, the subsequent formation of unstable cyclic polyselenides H, and the final thermal or oxidative selenium extrusion to result in the formation of relatively stable symmetrical 1,4-diselenins 4 and symmetrical 1,2,5-triselenepins 5. Though we could not isolate their isomeric products, 4′ and 5′, these compounds would also be formed as the minor components through this pathway and would be contained in the uncharacterized mixture of selenium-containing products. 1,3,5-Trithiones are generally recognized as the synthetic precursors of 1,6,6aλ4-trithiapentalenes.38-50 Therefore, it was assumed that the precursor of 1,6,6aλ4-triselenapentalene 6 should be 1,3,5-triselone or its alkeneselenolate-type intermediates J formed through homologation of borneneselenolate ion B at the C-3 position. Actually, treatment of compound 1 with t-BuOK and elemental selenium in HMPA afforded only 2, 3, and 5, and neither 6, 7, nor 8 were found at all in the crude products as shown in Table 1. This result strongly suggests that DMF, used as the solvent of the reactions, behaves as the C1 source for the formation of 6-8. Formation of selenocarbamate ions (I) by treating DMF with a base and elemental selenium has been reported previously,15,65-69 and, therefore, it was assumed that formylation or selenoformylation at the C-3 position of the bornane skeleton70,71 would proceed through the reaction of alkeneselenolate ion B with selenocarbamate ion (I) or its related derivatives under the strong basic conditions to form 1,3,5-triselenoxo-type intermediate J72 as shown in Scheme 6. However, addition of C1 sources, such as potassium carbonate (K2CO3), dimethyl carbonate, or CHCl3 in the reaction mixture did not affect the results at all, and introduction of CO2 gas to the reaction atmosphere only showed a slight improvement on the combined yield of 6-8. All the results of the attempts for optimization of the reaction condition are summarized in Table 1. The formation mechanism of unsymmetrical 4H-selenopyran-4-selone 8 also remained unclear at this time, but 8 was obtained as the minor component in all cases in contrast to 7, and 8 was assumed to be formed through a different pathway involving the reaction of 2-bornene or its carbene-type precursor generated in situ through Bamford–Stevens reaction of substrate 1.73
Plausible pathway for the formation of 4-7.
Reaction of d-Camphor p-Toluenesulfonylhydrazone (1) with t-BuOK and Elemental Selenium.
cA DMF solution of 1 was treated with t-BuOK and elemental selenium under an argon atmosphere and then dry CO2 gas was introduced to the reaction atmosphere along with an additional heating for 1.5 hours.
In conclusion, we found a new method for the synthesis of 1,6,6αλ4-triselenapentalene (6) and 4H-selenopyran-4-selones (7 and 8). Especially, our new findings on the short-step preparation of sterically crowded 1,6,6αλ4-triselenapentalene (6) are recognized to be synthetically useful by providing us a new motif of potential redox-permeable selenium compounds for the practical uses such as the components of organic electronic devices, and further studies on the reactivity of selenurane 6 involving their redox behaviors are expected in our laboratory.
Experimental
Instruments
The melting points were determined with a Büchi 535 micro melting point apparatus. 1H NMR spectra were recorded on a Bruker AC-400P (400 MHz) spectrometer, and the chemical shifts of the 1H NMR spectra are given in δ relative to internal tetramethylsilane. 13C NMR spectra were recorded on a Bruker AC-400P (100 MHz). 77Se NMR spectra were recorded on a Bruker AC-400P (76 MHz). Mass spectra were recorded on a Hitachi M-2000 mass spectrometer with electron-impact ionization at 20 or 70 eV using a direct inlet system. IR spectra were recorded for thin film (neat) or KBr disks on a JASCO FT/IR-7300 spectrometer. Elemental analyses were performed using a Yanagimoto CHN corder MT-5.
Materials
Column chromatography was performed using silica gel (Merck, Cat. No. 7734 or 9385) without pretreatment. Dichloromethane and chloroform were dried over phosphorus pentoxide and were freshly distilled before use. N,N-DMF and HMPA were dried over calcium hydride and were freshly distilled before use. Ethanol was dried over magnesium oxide and was freshly distilled before use. All substrates and reagents including cyclopentanone, d-camphor, p-toluenesulfonylhydrazide, acetic acid, potassium t-BuOK, elemental sulfur, elemental selenium, anhydrous potassium carbonate (K2CO3), anhydrous sodium sulfate (Na2SO4), sodium thiosulfate (Na2S2O3), NaBH4, and mCPBA were commercially available reagent grade and were used without any pretreatment.
General Procedure for the Reaction of Ketone p-Toluenesulfonylhydrazones with Potassium t-Butoxide and Elemental Selenium
A 30 mL DMF solution of p-toluenesulfonylhydrazone 1 (5.00 mmol) was treated with elemental selenium (1.00 g, 12.5 mmol) and potassium t-butoxide (1.40 g, 12.5 mmol) at room temperature, and the reaction mixture was heated at 100°C-150°C under an argon atmosphere for several hours. The reaction was then cooled to room temperature and was exposed to air for a few hours. The reaction was quenched with water and extracted with benzene. The organic layer was washed with water and dried over anhydrous Na2SO4 powder. After removing the solvent in vacuo, the crude product was purified using column chromatography on silica gel to afford dialkenyl diselenide 2, E/Z mixture of dimeric olefins 3, and several minor components (4, 5, 6, 7, and 8).
Physical and Spectral Data for Dialkenyl Diselenide 218
Colorless prism, monoclinic, P21 (#4), a = 7.857(3) Å, b = 14.637(5) Å, c = 9.005(4) Å, β = 97.09(3)°, V = 1027.7(6) Å3, Z = 2, Dcalc = 1.633 g/cm3, μ(MoKα) = 53.68 cm−1, R = 0.025, RW = 0.026. The data were deposited in Crystallographic Data Center (CCDC-1954439).
Red needle, monoclinic, P21 (#4), a = 15.668(1) Å, b = 8.3478(8) Å, c = 16.072(3) Å, β = 96.371(3)°, V = 2089.0(4) Å3, Z = 4, Dcalc = 1.607 g/cm3, μ(MoKα) =52.84 cm−1, R = 0.045, RW = 0.049. The data were deposited in Cambridge Crystallographic Data Center (CCDC-1954440).
Green platelet, orthorhombic, P212121 (#19), a = 6.779(3) Å, b = 13.334(4) Å, c = 21.610(7) Å, V = 1953(1) Å3, Z = 4, Dcalc = 1.491 g/cm3, μ(MoKα) =37.85 cm−1, R = 0.061, RW = 0.027. The data were deposited in Cambridge Crystallographic Data Center (CCDC-1954441).
Procedure for mCPBA Oxidation of 1,6,6aλ4-Triselenapentalene 6
A 30 mL CHCl3 solution of 1,6,6aλ4-triselenapentalene 6 (52 mg, 0.1 mmol) was treated with mCPBA (70%, 74 mg, 0.30 mmol) at room temperature for 30 minutes. Then, the reaction was quenched by addition of an excess amount of saturated Na2S2O3 solution, and saturated NaHCO3 solution was added to the reaction mixture. The reaction mixture was extracted with CH2Cl2, and the organic layer was washed with water and dried over anhydrous Na2SO4 powder. After removing the solvent in vacuo, the crude product was purified using column chromatography on silica gel to afford 1,2-diselenole 11 (37 mg, 81% yield) as orange crystals along with 4H-selenopyran-4-one 10 (trace) as a pale yellow oil.
General Procedure for Aerobic Oxidation of 4H-Selenopyran-4-Selones (7, 8)
A CHCl3 solution of symmetrical or unsymmetrical 4H-selenopyran-4-selone (7 or 8, 1.00 mmol) was standing at room temperature for 1 week, and the reaction mixture was filtered to remove the elemental selenium. Then, the solvent of the filtrate was removed invacuo. The crude product was purified using column chromatography on silica gel to afford the corresponding 4H-selenopyran-4-one 9 or 10, respectively, in quantitative yields.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID ID
Kazuaki Shimada
References
1.
EvansMB.HigginsGMC.MooreCGet al. Occurrence of an allylic rearrangement during the conversion of a dialkenyl disulfide to dialkenyl monosulfide by triphenylphosphine. Chem Ind. 1960;1960:897.
2.
DalgaardL.LawessonS-O. A new photo-rearrangement of a divinyl-disulphide. Tetrahedron Lett. 1973;14(44):4319-4320.doi:10.1016/S0040-4039(01)87210-5
3.
LarssonFCV.BrandsmaL.LawessonS-O. Thermal rearrangements of divinyl disulfides. Recl. Trav. Chim.Pays-Bas. 1974;93(9-10):258-260.doi:10.1002/recl.19740930908
4.
AmosovaSV.NosyrevaVV.SigalovMV. Synthesis of dialkenyl disulfides with phase-transfer catalysis. ZhOrg Khim.1992;28(5):927-929.
5.
TrofimovBA.SokolyanskayaLV.SenningA. Divinyl disulfides: synthesis and properties. Sulfur Reports.1992;12(1):1-55.doi:10.1080/01961779208048777
6.
RayPC. Synthesis of two bisthiocamphors. Nature. 1936;138(3491):548.doi:10.1038/138548a0
7.
SalamaP.PoirierM.CaissieM. Synthesis of a 1,2-dithiin mediated by electrophilic reagents. Heterocycles. 1996;41(11):2481-2485.doi:10.3987/COM-95-7164
8.
SalamaP.PoirierM.MayaMdelRP.RobichaudJ.BenoitM. First photolysis of an enolizable 1,4-Dithioketone: photochemical study of Bis-thiocamphor. Synlett. 1996;1996(09):823-824.doi:10.1055/s-1996-5614
9.
SalamaP.PoirierM. Single step first synthesis of a 3,6-dihydro-1,2-dithiin from its parent 1,4-dithioketone via a stereospecific rearrangement. Tetrahedron. 1997;8(16):2757-2760.doi:10.1016/S0957-4166(97)00279-6
10.
TestaferriL.TieccoM.TingoliM.ChianelliD. Stereospecific synthesis of divinyl diselenides from vinyl acetyl selenides. Tetrahedron. 1986;42(16):4577-4584.doi:10.1016/S0040-4020(01)87301-4
11.
DabdoubMJ.ComassetoJV. Divinyl ditellurides: synthesis and reactivity. J Organomet Chem. 1988;344(2):167-173.doi:10.1016/0022-328X(88)80476-5
12.
AmosovaSV.Gostevskaya VIGGM.PotapovVA.KashikAS. Preparation of divinyl ditelluride. Zh OrgKhim. 1988;24(2):454-455.
13.
AmosovaSV.PotapovVA.BulakhovaZA.RomanenkoLS. 1,4-Ditellurin and 2-methylene-1,3-ditellurole from tellurium metal and acetylene. Sulfur Lett. 1991;13(4):143-146.
14.
HuangX.WangJ-H. A stereoselective synthesis of (E)-divinyl diselenides and (E)-divinyl ditellurides. SynthCommun.2000;30(2):301-306.doi:10.1080/00397910008087322
15.
ShimadaK.OikawaS.NakamuraHet al. A preparation of alkyl or alkenyl N,N-dimethyl chalcogenocarbamates and their one-step conversion into symmetrical dialkyl or dialkenyl dichalcogenides. BullChem Soc Jpn. 2005;78(5):899-905.doi:10.1246/bcsj.78.899
16.
MusalovaMV.PotapovVA.PanovVA.AmosovaSV. Synthesis of divinyl ditelluride from tellurium and acetylene. Russ J Org Chem. 2012;48(5):743-744.doi:10.1134/S107042801205020X
17.
MuraiT. Generation and reactions of selenium isologues of enolate ions. Mini-Reviews in OrganicChemistry. 2004;1(3):279-290.doi:10.2174/1570193043403208
18.
ShimadaK.AsahidaM.TakahashiKet al. Synthesis of dialkenyl dichalcogenides via alkenechalcogenolate ions generated by treating ketone p-toluenesulfonylhydrazones with a base and elemental chalcogen. ChemLett. 1998;27(6):513-514.doi:10.1246/cl.1998.513
19.
TakikawaY.UwanoA.WatanabeH.AsanumaM.ShimadaK. Novel synthesis of 1,3,5-triselenanes from aldehydes, and novel generation of selenoaldehydes by fragmentation of 1,3,5-triselenanes. Tetrahedron Lett. 1989;30(44):6047-6050.doi:10.1016/S0040-4039(01)93851-1
20.
ShimadaK.JinN.FujimuraM.NaganoY.KudohE.TakikawaY. An efficient synthesis of selenocarbonyl compounds by the treatment of carbonyl compounds with bis[1,5-cyclooctanediyl]boryl selenide. Chem Lett. 1992;21(9):1843-1846.doi:10.1246/cl.1992.1843
21.
ShimadaK.JinN.KawaguchiMet al. Efficient synthesis of selenocarbonyl compounds by treating carbonyl compounds with bis(1,5-cyclooctanediylboryl) selenide. Bull Chem Soc Jpn. 1997;70(1):197-206.doi:10.1246/bcsj.70.197
22.
ShimadaK.GongY.NakamuraH.MatsumotoR.AoyagiS.TakikawaY. Novel generation of selenoaldehydes through stannic chloride-Induced unsymmetrical C-Se bond cleavage of bis(N,N-dimethylcarbamoylseleno)methanes. Tetrahedron Lett. 2005;46(21):3775-3778.doi:10.1016/j.tetlet.2005.03.153
23.
GongY.ShimadaK.NakamuraHet al. Stannic chloride-induced unsymmetrical C-Se bond cleavage of bis(N,N-dimethylcarbamoylseleno)methanes: Novel generation of selenoaldehydes. Heteroatom Chem. 2006;17(2):125-135.doi:10.1002/hc.20190
24.
ShimadaK.IzumiH.OtashiroKet al. A novel one-step synthesis of quinoline-2(1H)-thiones and selones by treating 3-aryl-3-(2-aminophenyl)-1-propyn-3-ols with a base and sulfur or selenium. NatProdCommun. 2015;10(6):903-912.doi:10.1177/1934578X1501000628
25.
ClarkeP.WhitingMC.PapenmeierG.ReuschW. Solvent effects in the decomposition of diazocamphane. JOrg Chem.1962;27(9):3356-3357.doi:10.1021/jo01056a532
26.
ShapiroRH.HeathMJ. Reaction of tosylhydrazones with alkyllithium reagents. A new olefin synthesis. JAmChem Soc. 1967;89(22):5734-5735.doi:10.1021/ja00998a601
ChandrasekharS.RajaiahG.ChandraiahL.SwamyDN. Direct conversion of tosylhydrazones to tert-butyl ethers under Bamford-Stevens reaction conditions. Synlett. 2001;2001(11):1779-1780.doi:10.1055/s-2001-18083
29.
TothM.SomsakL. exo-Glycals from glycosyl cyanides. First generation of C-glycosylmethylene carbenes from 2,5- and 2,6-anhydroaldose tosylhydrazones. J Chem Soc Perkin Trans1. 2001;2001(9):942-943.
30.
FultonJR.AggarwalVK.de VicenteJ. The use of tosylhydrazone salts as a safe alternative for handling diazo compounds and their applications in organic synthesis. EuropeanJ Org Chem. 2005;2005(8):1479-1492.doi:10.1002/ejoc.200400700
31.
MeichsnerE.NierengartenI.HollerM.ChesséM.NierengartenJ-F. A fullerene-substituted pillar[5]arene for the construction of a photoactive rotaxane. Helv Chim Acta. 2018;101(6):e1800059.doi:10.1002/hlca.201800059
32.
HockKJ.KoenigsRM. The generation of diazo compounds in continuous-flow. Chem - A Eur J. 2018;24(42):10571-10583.doi:10.1002/chem.201800136
33.
MeteTB.LahaD.BhatRG. A one pot transition-metal-free synthesis of styrenyl ethers from 2-aryloxy/alkoxy acetophenones. ChemistrySelect.2018;3(26):7656-7659.doi:10.1002/slct.201801522
34.
OppolzerW. Camphor derivatives as chiral auxiliaries in asymmetric synthesis. Tetrahedron. 1987;43(9):1969-2004.doi:10.1016/S0040-4020(01)86780-6
35.
ChelucciG. Synthesis and application in asymmetric catalysis of camphor-based pyridine ligands. Chem SocRev. 2006;35(12):1230-1243.doi:10.1039/b604793a
GroseljU. Camphor-derivatives in asymmetric organocatalysis - Synthesis and application. Curr Org Chem.2015;19(21):2048-2074.doi:10.2174/1385272819666150713180204
38.
Lozac’hN.VialleJ. Chemistry of the 1,2-dithiole ring. Organic Sulfur Compounds. 1996;2:257-285.
39.
ClarkDT. Structure and bonding in organosulfur compounds. International Journal of Sulfur Chemistry,Part C: Mechanisms of Reactions of Sulfur Compounds. 1972;7:11-32.
40.
HordvikA.SaethreLJ. Structure studies on 6a-thiathiophthenes and related compounds. Isr J Chem. 1972;10(2):239-248.doi:10.1002/ijch.197200030
41.
BeerRJS. 6a-Thiathiophthenes and related compounds. Organic Compounds of Sulphur, Selenium, andTellurium. 1973.;2:497-510.
42.
BeerRJS. 6a-Thiathiophthenes and related compounds. Organic Compounds of Sulphur, Selenium, andTellurium. 1975;3:495-508.
43.
GleiterR.GygaxR. No-bond-resonance compounds, structure, bonding and properties. Top Curr Chem. 1976;63:49-88.
44.
BeerRJS. 6a-Thiathiophthenes and related compounds. Organic Compounds of Sulphur, Selenium, andTellurium. 1977;4:300-307.
45.
NaganoT. Enhancers of superoxide generation in vivo and novel superoxide dismutase mimics. J Syn OrgChem Jpn. 1989;47(9):843-854.doi:10.5059/yukigoseikyokaishi.47.843
46.
PedersenCT. Product class 7: 1,2-Dithiolium salts and related compounds. Science of Synthesis.2002;11:107-189.
47.
AkibaK.YamamotoY. Dynamic aspects of hypervalent compounds effected by the formation of three center-four electron bond in heteroatoms. Heteroatom Chem. 2007;18(2):161-175.doi:10.1002/hc.20326
48.
RašovićA.KochA.KleinpeterE.MarkovićR. Studies of the regioselective ring-opening–closing mode of functionally different thiazolidine type enaminones: en route to the synthesis of trithiaazapentalene derivatives. Tetrahedron. 2013;69(51):10849-10857.doi:10.1016/j.tet.2013.10.088
49.
AttaKFM.FarahatOOM.Al-ShargabiTQ.MareiMG.El AshryESH. Chemistry of pent-4-yne-1,3-diones (acetylenic β-diketones) as precursors for heterocyclic compounds. Advances in Heterocyclic Chemistry. 2014;113:67-110.
50.
GleiterR.HaberhauerG. Electron-rich two-, three- and four-center bonds between chalcogens – new prospects for old molecules. Coord Chem Rev. 2017;344:263-298.doi:10.1016/j.ccr.2017.03.003
51.
HordvikA.JulshamnK.KihlborgL.BorchG.SchaumburgK.EhrenbergL. The crystal and molecular structure of 6a-selenaselenophthene. Acta ChemScand. 1971;25(7):2507-2515.doi:10.3891/acta.chem.scand.25-2507
52.
HordvikA.PortenJA.HebrewC.Van BurenCT.KlæboeP.SwahnC-G. The crystal and molecular structure of 3,4-trimethylene-6a-selenaselenophthene. Acta Chem Scand. 1973;27(2):485-492.doi:10.3891/acta.chem.scand.27-0485
53.
MoellerJ.ChristieRM.PedersenCT.ReidDH. Mass spectrometric studies of selenaanalogs of 1,6,6aλ4-trithiapentalenes (1,6,6aλ4-triselenapentalenes and 1,6,6aλ4-diselena-5,6-diazapentalenes. Acta Chem ScandSer B: Organic Chemistry and Biochemistry. 1976.;B30(7):600-604.
54.
SaethreLJ.MalmquistPA.MartenssonN.SvenssonS.GeliusU.SiegbahnK. Solid-state ESCA studies of trithiapentalene and selenium analogs. Inorg Chem. 1981;20(2):399-402.doi:10.1021/ic50216a016
55.
BhattacharyyaP.SlawinAMZ.WoollinsJD. Reaction of [{PhP(Se)(µ-Se)}2] with dialkyl cyanamides: X-ray crystal structures of the phosphorus-containing triselenapentalenes [Me2N-C(Se)=N]2P(Se)Ph and [O(CH2CH2)2N-C(Se)=N]2P(Se)Ph. Angew Chem Int Edit.2000;39(11):1973-1975.doi:10.1002/1521-3773(20000602)39:11<1973::AID-ANIE1973>3.0.CO;2-U
56.
HordvikA.JulshamnK.SeppäläIJ.HedbergK.SchaumburgK.EhrenbergL. The structure of 6a-selenathiophthene. Acta Chem Scand. 1971;25(5):1895-1896.doi:10.3891/acta.chem.scand.25-1895
DingY.KongJ.ReidDH. Studies of heterocyclic compounds. Part 36 Trithia- and dithiaselenapentalenes from benzylidene-1,2-dithioles and heterocumulenes. Heteroatom Chem. 1997;8(3):233-244.
59.
WynbergH.LammertsmaK.HulshofLA. The synthesis and chiroptical properties of the two diastereomeric 2-bornanylidenebornanes. Tetrahedron Lett. 1975;16(43):3749-3752.doi:10.1016/S0040-4039(00)91327-3
60.
TakeshitaH.HatsuiT.JinnaiO. Sensitized photooxidation of cis-, and trans-2-bornylidene-2-bornanes: a formation of an epoxycyclopropane derivative during the oxygenation. Chem Lett. 1976;5(10):1059-1062.doi:10.1246/cl.1976.1059
BlockE.BirringerM.DeOrazioRet al. Synthesis, properties, oxidation, and electrochemistry of 1,2-dichalcogenins. J Am Chem Soc. 2000;122(21):5052-5064.doi:10.1021/ja994134s
63.
DettyMR.LussHR. Addition of disodium chalcogenides to 1,5-bis(trimethylsilyl)penta-1,4-diyn-3-one. Synthesis, structure, and reactivity of the parent δ-4H-chalcogenapyran-4-ones. Organometallics. 1992;11(6):2157-2162.doi:10.1021/om00042a032
64.
LeonardK.NelenM.RaghuM.DettyMR. Chalcogenapyranones from disodium chalcogenide addition to 1,4-pentadiyn-3-ones. The role of enol ethers as intermediates. J Heterocycl Chem.1999;36(3):707-717.doi:10.1002/jhet.5570360322
65.
MizunoT. New carbonylation using an unstable intermediate. Kagaku to Kogyo.1995;69(6):240-248.
66.
NudelmanNS.SchulzH.LinaresGG.BonattiA.BocheG. Further insights into the chemistry of acyllithium compounds R2NC(O)Li; Characterization of an amide (R2NLi) adduct (R2NCHNR2(OLi)) to a formamide (R2NC(O)H). Organometallics. 1998;17(2):146-150.
67.
ReevesJT.TanZ.HerbageMAet al. Carbamoyl anion addition to N-sulfinyl imines: Highly diastereoselective synthesis of α-amino amides. J Am Chem Soc. 2013;135(15):5565-5568.doi:10.1021/ja402647m
SeifertCW.PindiS.LiG. Asymmetric carbamoyl anion additions to chiral N-Phosphonyl imines via the gap chemistry process and stereoselectivity enrichments. J Org Chem. 2015;80(1):447-452.doi:10.1021/jo5024443
70.
BackTG.DyckBP.ParvezM. Unexpected formation of 1,3-diselenetanes from the reaction of camphor enolate with selenium. J Chem Soc Chem Commun. 1994;1994(4):515-516.doi:10.1039/c39940000515
71.
BackTG.DyckBP.ParvezM. 1,3-Diselenetanes and 1,3-dithietanes derived from camphor. Formation, structure, stereochemistry, and oxidation to selenoxide and sulfoxide product. J Org Chem. 1995;60(3):703-710.doi:10.1021/jo00108a038
72.
ReidDH.WebsterRG. Studies of heterocyclic compounds. Part XII. A three-step synthesis of 6a-thiathiophthen from γ-pyrone. J. Chem. Soc., Perkin Trans. 1. 1972;1972:1447-1449.doi:10.1039/P19720001447
73.
KernerJA.NickonA. Hydrogen shifts in cyclohexylcarbenes. Spatial dependence of activating power and of primary deuterium isotope effects. Tetrahedron. 1997;53(44):14871-14894.