
Abstract
Starting from an unprotected GlcNAc acceptor, after first random glycosylation and deprotection, and second random glycosylation, deprotection, and purification, we were able to obtain a trisaccharide library containing 36 α-linked trisaccharide library. These compounds were characterized by 1D, 2D NMR and mass spectra. The library was used as the starting material for enzymatic reaction to obtain a tetrasaccharide in support of the results. Based on this method, a trisaccharide library containing 15 α-linked trisaccharides was synthesized.
Glycoconjugates play an important role in cellular recognition, adhesion, cell-growth regulation, cancer cell metastasis, and inflammation. 1,2 The carbohydrates on the cell surface serve as anchor sites for infectious bacteria, viruses, and toxins. 3 Therefore, synthesis of novel carbohydrates and their screening against disease causing agents may help in finding the inhibitors of some critical pathogenic processes. However, chemical synthesis of carbohydrates is laborious, time consuming, and technically demanding. 4 There is a large scope to find more efficient methods for generating the oligosaccharide libraries.
Random glycosylation strategy was demonstrated as an excellent combinatorial synthetic method for generating oligosaccharide library.
5,6
This strategy eliminated the need for numerous synthetic steps to construct orthogonally protected sugar building blocks and reduced both the time and the cost of synthesis. For example, a trisaccharide library was obtained (Scheme 1) from random fucosylation of per-O-benzylated fucosyl trichloroacetimidate
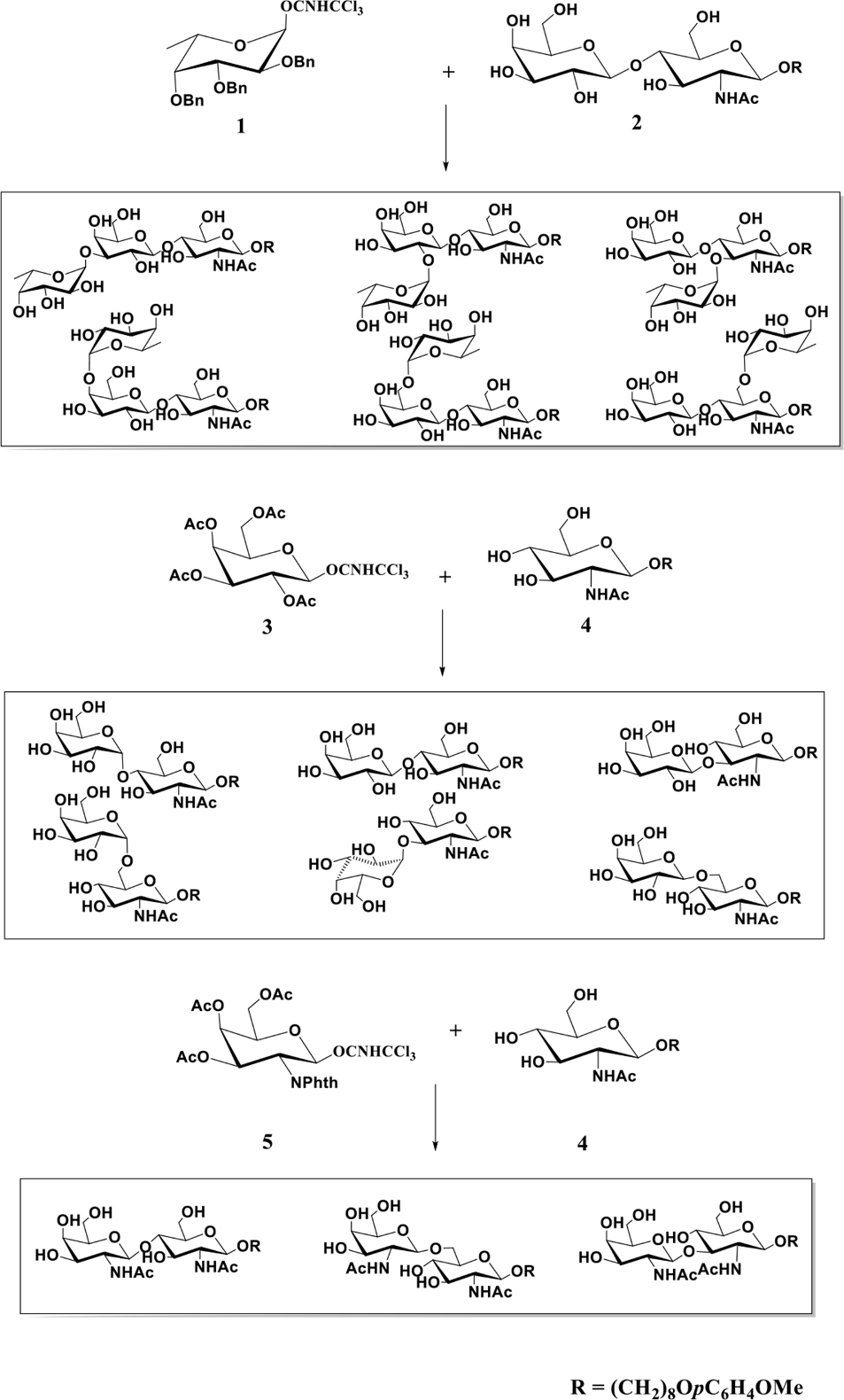
Synthesis of disaccharide and trisaccharide libraries from random glycosylation.
In continuation of our interest in random glycosylation, we have been interested to double this method in the synthesis of complex libraries of oligosaccharides. We believe that this double random glycosylation will allow us to synthesize more complex oligosaccharide libraries in an efficient manner. In this communication, we report our findings on the synthesis of oligosaccharide libraries through double random glycosylations.
Results and Discussion
The target library we like to synthesize through double random glycosylation strategy was listed in Figure 1, and its synthetic strategy was summarized in Scheme 2.
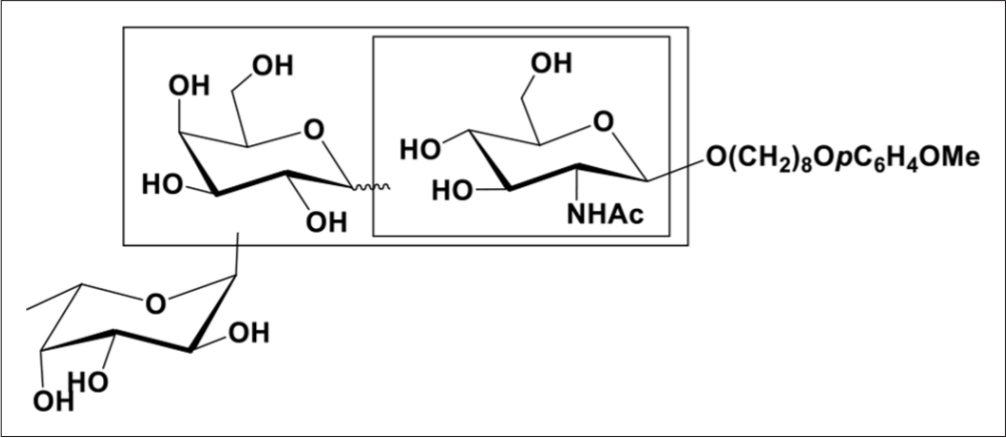
The target oligosaccharide library containing 36 trisaccharides.

Reagents and conditions: (a) i: dioxane, BF3·Et2O, room temperature; ii: H2, Pd/C. (b) i: dioxane, TMSOTf, room temperature; ii: NaOMe/MeOH.
Accordingly, synthesis of the target library started from the unprotected GlcNAc acceptor
Compound
Theoretically, the mixture should contain 36 α-linked fucosylated trisaccharides, and its 1D proton nuclear magnetic resonance (NMR) spectrum was very complex and could not derive any positive information to support the results (as shown in Supplemental Figures S-1 and S-2).
However, its 2D NMR spectra (Figures 2 and 3) were more forthcoming in delineating the results. Thus, 36 peaks were observed from 5.4 to 4.7 ppm, which correspond to H-1 in fucosyl unit in trisaccharides; the peaks from 2.1 to 2.9 ppm, correspond to H-5 protons in fucosyl unit which unequivocally confirm the formation of desired fucosylated trisaccharides.
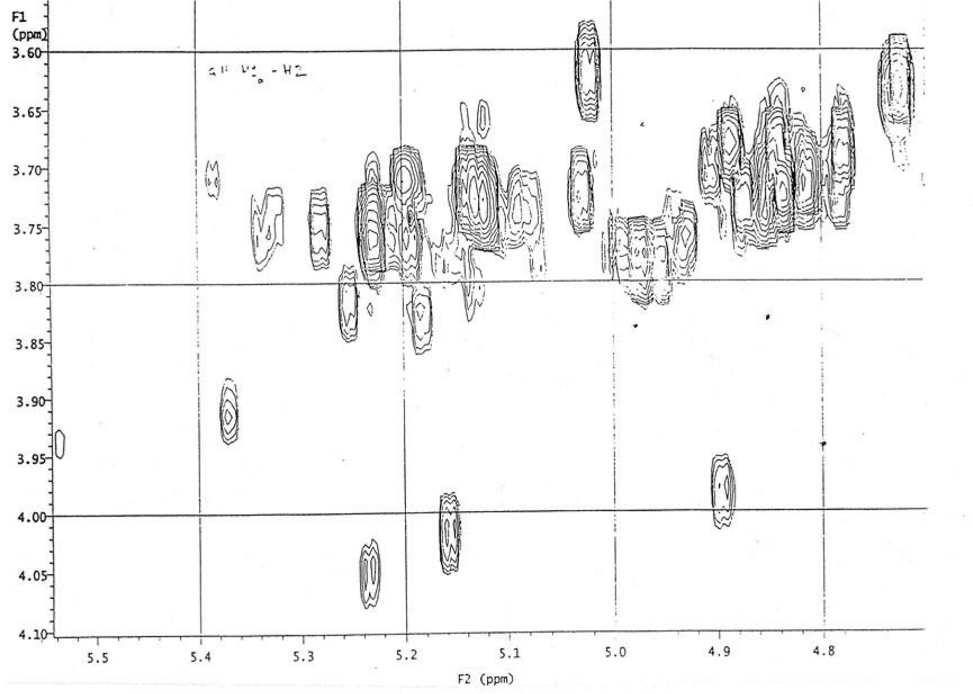
2D NMR spectra of trisaccharide library from the double random glycosylations (as shown in Supplemental Figure S-3).
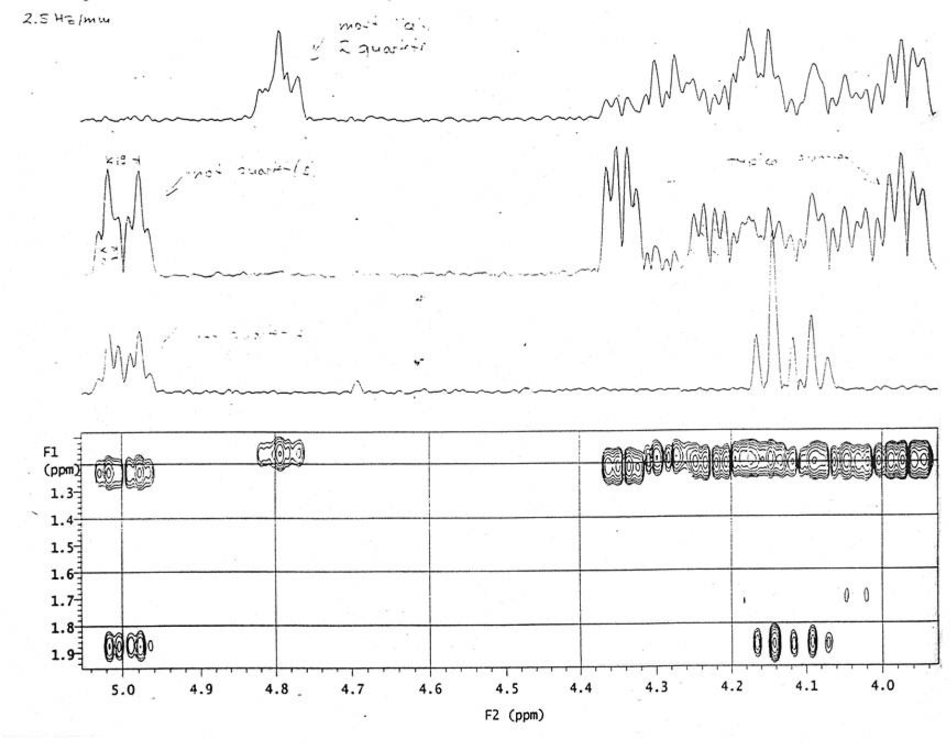
2D NMR spectra of trisaccharide library from the double random glycosylations (as shown in Supplemental Figure S-4).
A further confirmation of this library could be drawn from the selective enzymatic galactosylation. Accordingly, the mixture having 6 trisaccharides containing 2-α-fucosyl galactosyl fragment acts as the galactosylation acceptor for enzymatic α-galactosylation with UDP-gal in the presence of blood B transferase. Thus, the double random glycosylation products in 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 7.0) was treated with recombinant blood B α-1,3-galactosyltransferase in the presence of 5 µmol of UDP-Gal and 5 µmol of MnCl2 at 37°C. After 24 hours incubation, more enzyme and UDP-Gal were added, and the mixture was incubated for another 24 hours. The mixture was then purified on C-18 column to give the enzymatic α-galactosylation products. The mass spectrum of this mixture indicated the formation of tetrasaccharides (Scheme 3).
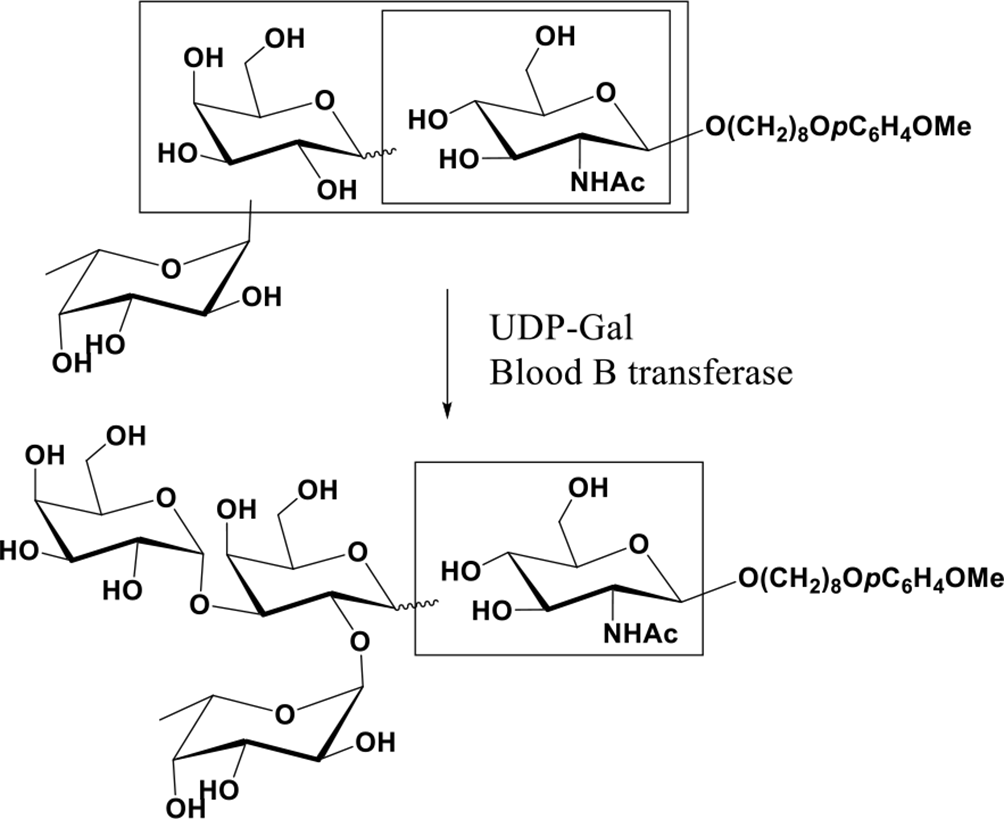
Enzymatic α-galactosylation on the double random glycosylation products.
The comparison of HPLC chromatograms of the starting materials (Figure 4) with that of the enzymatic reaction mixture is given in Figure 5. A critical analysis of the chromatograms clearly reveals that 6 additional compounds were formed during the enzymatic reaction. However, it was extremely hard to isolate all of 6 tetrasaccharides through C-18 column or HPLC due to the small percentages of the tetrasaccharides. Based on the HPLC chromatogram of the enzymatic products, it is to be observed that one of the compounds was relatively in higher percentage in comparison to others. Therefore, through repeated C-18 column purification, we were able to obtain a compound whose mass spectrum confirmed that it was a tetrasaccharide, and its proton NMR spectrum (Figure 6 as shown in Supplemental Figures S-7 and S-8) indicated that there are 2 β linkages and 2 α linkages in the compound. This tetrasaccharide should be β (1-4) Gal-GlcNAc, or β (1-3) Gal-GlcNAc or β (1-6) Gal-GlcNAc related tetrasaccharide.
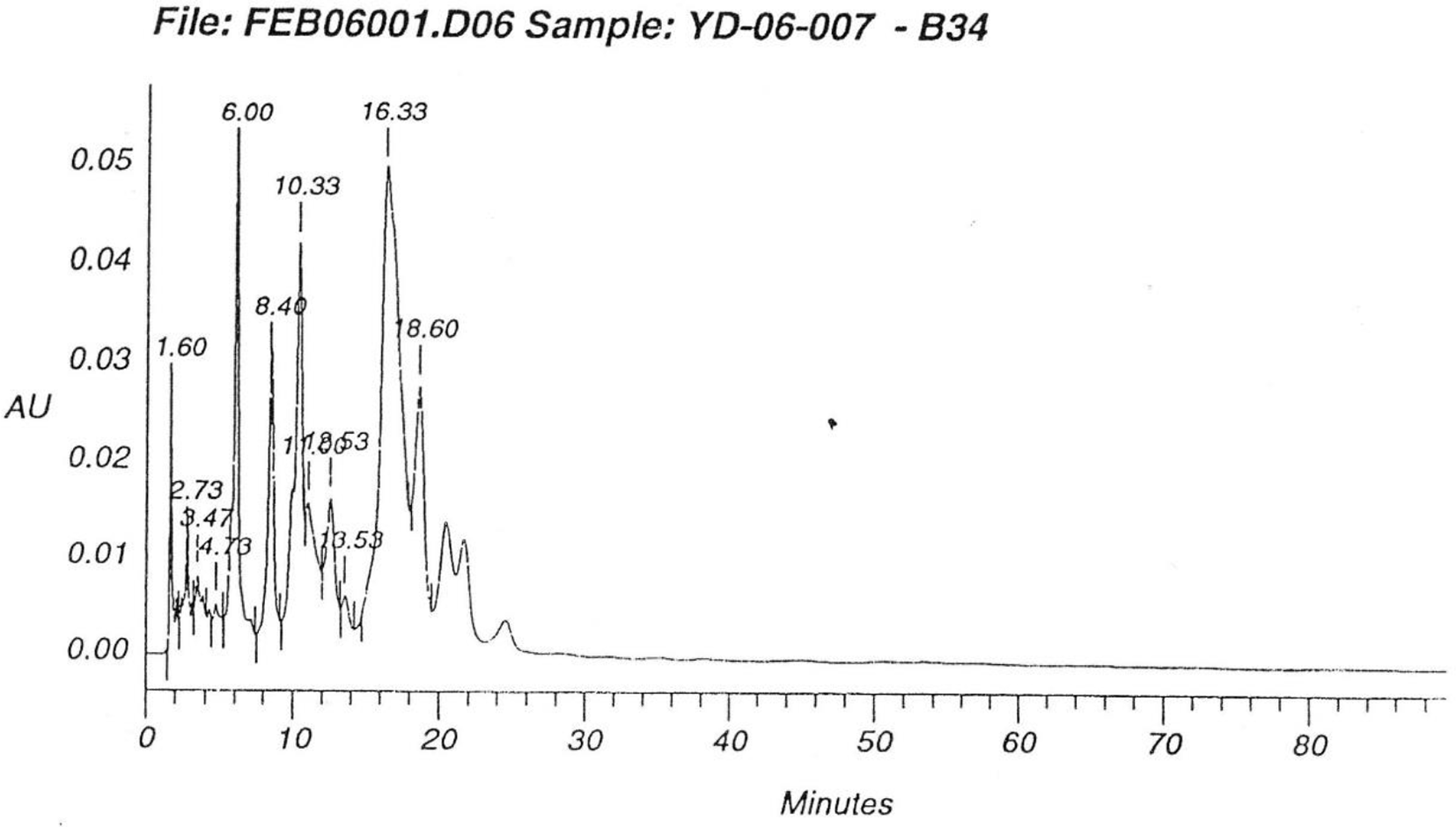
The HPLC chromatograms of the trisaccharide library from double random glycosylations (as shown in Supplemental Figure S-5).

The HPLC chromatograms of the starting materials and the products of the enzymatic reaction (as shown in Supplemental Figure S-6).
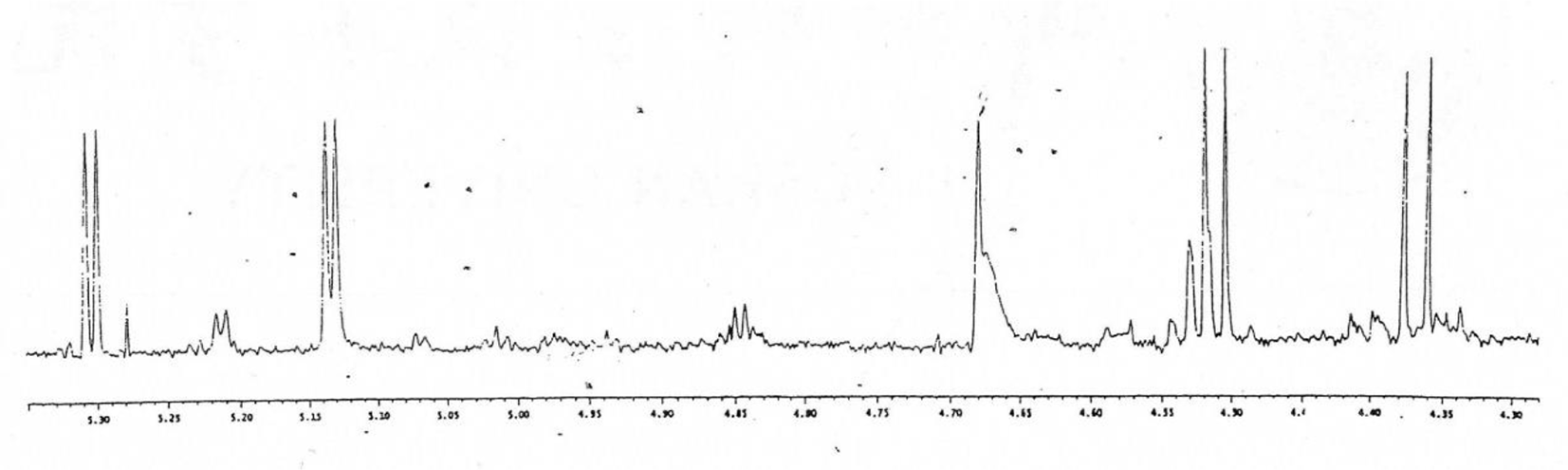
The 1H-NMR spectrum of the tetrasaccharide from the enzymatic α-galactosylation reaction (as shown in Supplemental Figure S-8).
It is very gratifying to note the successful completion of design and execution of target double random glycosylation library. As an extension of this approach, we used this strategy for the synthesis of another oligosaccharide library. Thus, GalNAc-GlcNAc related trisaccharide library was designed and synthesized through double random glycosylation strategy as shown in Scheme 4.

Synthesis of β-linked GalNAc-GlcNAc related library through double random glycosylation.
Thus, the unprotected GlcNAc acceptor

The 1H-NMR spectrum of GalNAc-GlcNAc related trisaccharide library through double random glycosylation (as shown in Supplemental Figure S-10).
This library should contain 15 α-linked GalNAc-GlcNAc trisaccharides, the proton NMR spectrum should display 15 peaks for H-1 in fucosyl units from 5.10 to 4.69 ppm, and we can see 15 peaks in this range.
Conclusion
In conclusion, a novel double random glycosylation strategy is developed for efficient synthesis of complex oligosaccharide library. Gratifyingly, the results were confirmed by a combination of NMR and mass spectral studies followed by enzymatic α-galactosylation. We believe that the double random glycosylation strategy described here is thus far the most efficient method for the synthesis of complex oligosaccharide libraries.
Experimental Part
TLC was performed on silica gel 60-F254 and charred with H2SO4. 1H NMR spectra were recorded at 300 MHz (Bruker AM-300), 360 MHz (Bruker AM-360), or 500 MHz (Varian UNITY 500) in CDCI3 (internal TMS, δ 0), or D2O (internal acetone, δ 2.23) solution; 13C NMR spectra were recorded at 75 or 90 MHz on the same instruments in CDC13 (internal TMS, δ 0) or D2O (internal dioxane, δ 67.4) solution. MALDI MS was obtained from Kratos Kompact MALDI. Methanol was distilled over Mg, and toluene was distilled over CaH2.
Double Random Glycosylation Procedure
To a solution of disaccharide library from single random glycosylation in dry DMF was added powdered dry CaSO4 and 4 equivalents of 2,3,4-tri-O-benzyl fucosyl imidate (
Enzymatic Reaction Procedure
The solution of trisaccharide library (10 mg) in MES buffer (pH 7.0) was treated with recombinant rat liver α-1,3-galactosyltransferase in the presence of 5 µmol of UDP-Gal and 5 µmol of MnCl2 at 37°C. After 24 hours incubation, more enzyme and UDP-Gal were added, and the mixture was incubated for another 24 hours. After work up, the reaction mixture was purified on C-18 column to give the products. After purifications on silica gel column and C-18 column, 1 tetrasaccharide was obtained. 1H NMR (CD3OD, 300 MHz): δ 5.30 (d, 1H, J = 4.70 Hz), 5.14 (d, 1H, J = 4.78 Hz), 4.51 (d, 1H, J = 8.04 Hz), 4.37 (d, 1H, J = 4.70 Hz); ESIMS: m/z 675 [M+H]+.
Supplemental Material
Supplementary material - Supplemental material for Synthesis of Oligosaccharide Libraries Through Glycosylations on Unprotected Acceptors
Supplemental material, Supplementary material, for Synthesis of Oligosaccharide Libraries Through Glycosylations on Unprotected Acceptors by Yili Ding, Chamakura V. N. S. Vara Prasad and Bingyun Wang in Natural Product Communications
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.