Prenylated flavonoids have attracted much attention due to their promising and diverse bioactivities on multitarget tissues. To the best of our knowledge, our recent studies demonstrated first that (2S)-6-prenylnaringenin (6-PNG), a hop component, blocks Cav3.2 T-type calcium channels (T-channels) and alleviates neuropathic and visceral pain with little side effects; it also indicated first that other natural prenylflavanones (PFVNs), such as sophoraflavanone G and (2S)-8-PNG, or synthetic 6-PFVNs including (2R/S)-6-PNG and its derivatives are capable of blocking T-channels and useful for pain therapy. Through the structure-activity relationship studies on the synthetic 6-PFVNs, we identified 6-(3-ethylpent-2-enyl)-5,7-dihydroxy-2-(2-hydroxyphenyl)chroman-4-one (8j or KTt-45) as the most potent blocker of Cav3.2 T-channels. It is interesting to recognize a prenylated flavonoid, belonging to other sub-classes, as a novel T-channel blocker. Therefore, this article will review some of our recent studies to introduce a new branch to researchers studying on prenylated flavonoids.
Prenylated flavonoids are a sub-class of flavonoids, which combine a flavonoid skeleton with a lipophilic prenyl side-chain. About1000 prenylated flavonoids have been identified from plants. Prenylation has occurred on most of flavonoid skeletons, including chalcones, flavanones, flavones, isoflavones, and flavonols to yield the diverse sub-classes of prenylated flavonoids. The most common sub-class is prenylflavanones (PFVNs), while the rarest sub-class is prenylated flavanols. In general, the C-prenylated flavonoids are much more popular than the O-prenylated flavonoids.1 The prenyl side-chain generally refers to alkyl-substituent groups, such as prenyl, geranyl, and lavandulyl groups as well as the variations of these alkyl groups depending on their oxidation and hydroxylation.2 The prenylation may make the prenylated flavonoids have higher affinity with biological membranes and have better interaction with target proteins than their flavonoid skeletons. As a result, the prenylated flavonoids usually possess a wide variety of bioactivities, including estrogenic, antioxidant, immunomodulatory, anti-inflammatory, and anticancer activities. Thereby, the prenylated flavonoids have attracted much attention as novel nutraceuticals3 or as seeds for developing new drugs.4 This attraction of the prenylated flavonoids will rise since the common PFVNs, such as sophoraflavanone G (I or SG), (2S)-8-prenylnaringenin (II or (2S)-8-PNG), and (2S)-6-PNG (III), or the synthetic 6-PFVNs including (2 R/S)-6-PNG (IV) and its derivatives (Figure 1) have just been recognized as a novel class of T-type calcium channel (T-channel) blockers and these new T-channel blockers are useful for pain therapy.5,6 These results are likely to contribute greatly to the development of safer and more effective drugs for pain therapy. Currently, the drugs for pain therapy remain insufficient for certain forms of pain associated with chronic disorders (eg, neuropathic pain) and often have serious side effects.7 Therefore, this review will offer historical background information on recognizing PFVNs as novel T-channel blockers, using PFVNs to alleviate pain, and studying the structure-activity relationship (SAR) based on synthetic 6-PFVNs.
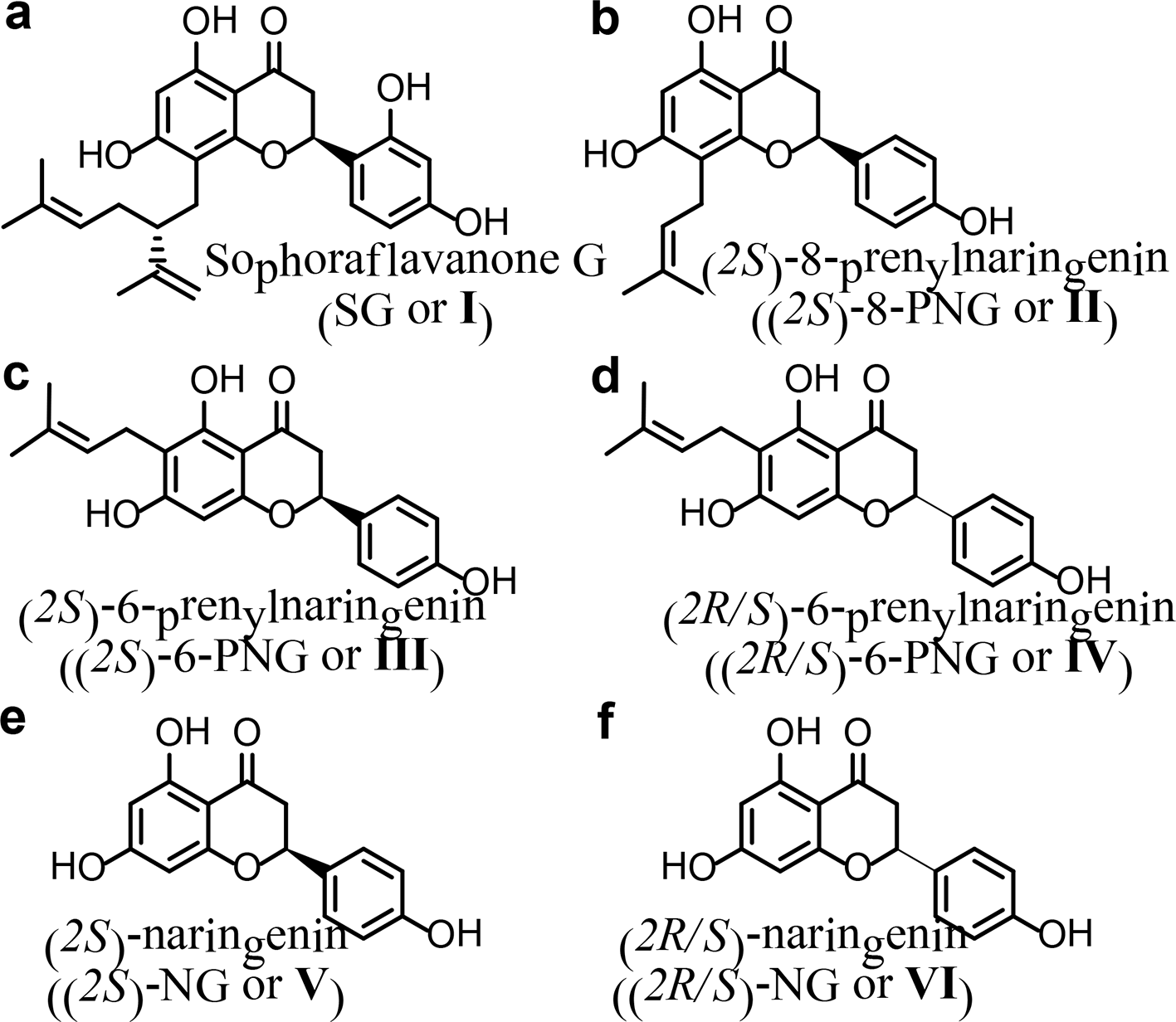
The structures of prenylflavanones including sophoraflavanone G (SG or I) (a), (2S)-8-prenylnaringenin ((2S)-8-PNG or II) (b), (2S)-6-prenylnaringenin ((2S)-6-PNG or III) (c), and (2R/S)-6-PNG (IV) (d), and the structures of (2S)-naringenin ((2S)-NG or V) (e) and (2R/S)-NG (VI) (f).
Recognizing PFVNs as Novel T-Type Calcium Channel Blockers
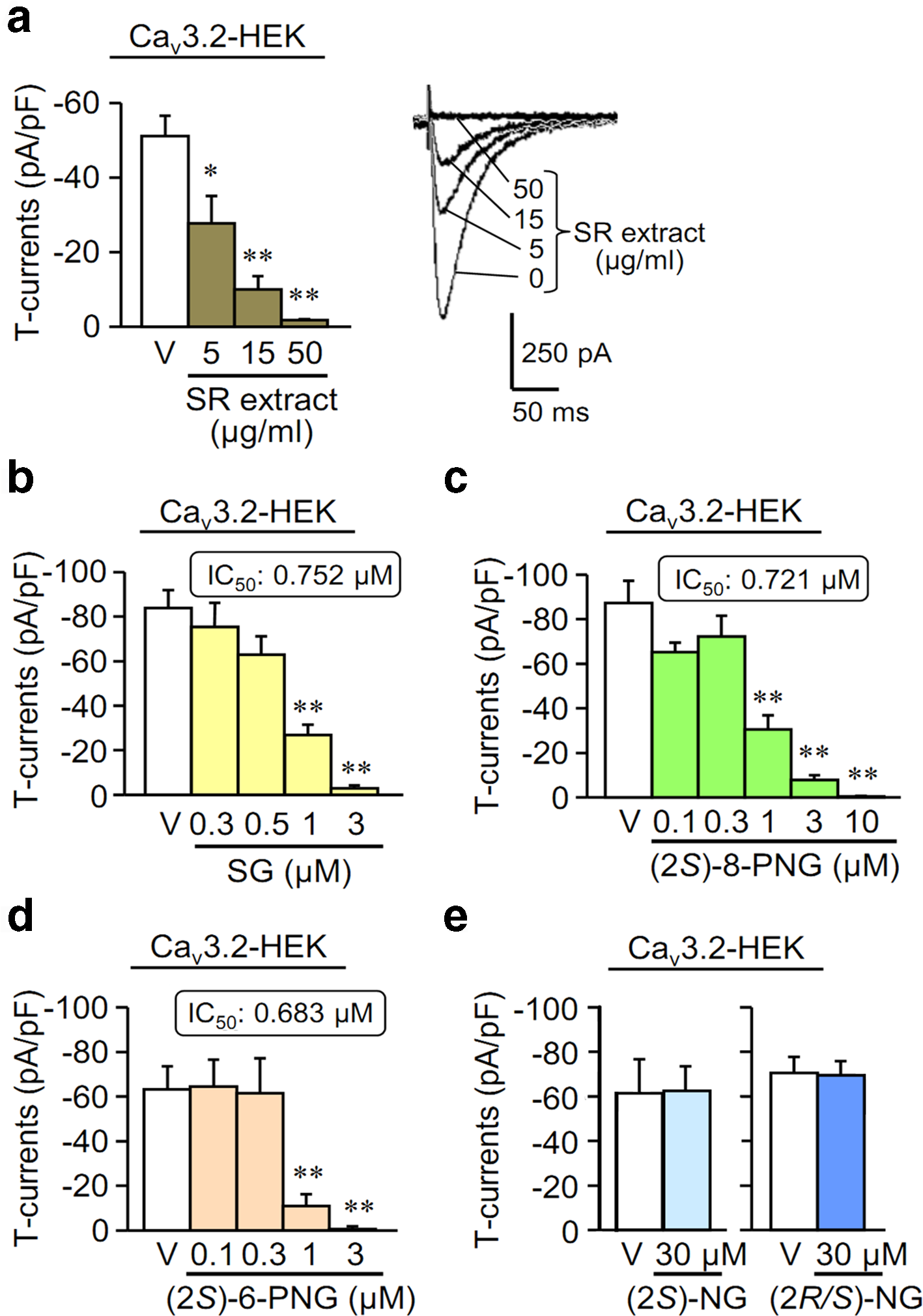
T-channels are expressed on the peripheral endings, central terminals and the dorsal root ganglion of primary sensory neurons, and the T-channels are also found in some parts of the brain such as thalamus.7-13 These organs involve in the pain pathway, so the T-channel blockers are more likely to alleviate pain. Hence, PFVNs were first recognized to be useful for pain therapy through the evaluation of T-channel-blocking,5 which will be reviewed herein. First of all, a 50% ethanol extract of sophorae radix (SR) was evaluated for the T-channel-blocking activity on human Cav3.2-expressing HEK293 (Cav3.2-HEK) cells. The result shows that the SR-extract inhibited T-channel-dependent currents (T-currents) in a range of 5 to 50 µg/mL (Figure 2(a)).5 After separation of the SR-extract by chromatography, the fraction exhibiting the most potent activity in a range of 0.3 to 1 µg/mL was determined.5 From this fraction, SG (I) was identified as an active compound (Figure 1(a)). I strongly inhibited T-currents in Cav3.2-HEK cells (Figure 2(a)) and Cav3.1-HEK cells, but exhibited only minor inhibitory effect on the high voltage-activated (HVA) Ca2+ channel-dependent currents (HVA-currents) in differentiated NG108-15 cells that express L-, N-, P/Q- and R-types of calcium channels.5
The inhibitory effects of the T-type Ca2+ channel-dependent currents (T-currents) in a concentration-dependent manner of the extract of sophorae radix (SR-extract) (a), sophoraflavanone G (I or SG) (b), (2S)-8-prenylnaringenin (II or (2S)-8-PNG) (c), (2S)-6-prenylnaringenin (III or (2S)-6-PNG) (d), (2S)-naringenin (V or (2S)-NG) (e) and (2R/S)-NG (VI) (e) in HEK293 cells, transfected with human Cav3.2 T-channels (Cav3.2-HEK). Data show the mean ± SEM for 5 to 9 (a), 5 to 18 (b), 6 to 13 (c), 5 to 8 (d), and 5 to 13 (e) experiments. *P < 0.05, **P < 0.01 vs vehicle (V).5
Next, the T-channel-blocking activity of simpler PFVNs including (2S)-8-PNG (II) (Figure 1(b)) and (2S)-6-PNG (III) (Figure 1(c)), as well as their basic skeleton structure, (2S)-naringenin ((2S)-NG or V) and (2R/S)-NG (VI) (Figure 1(e) and (f)), was evaluated in Cav3.1-HEK and Cav3.2-HEK cells. II and III also suppressed T-currents in Cav3.2-HEK (Figure 2(c) and (d)) and Cav3.1-HEK cells, comparable to I. The IC50 proportion of Cav3.1 to Cav3.2 T-type channel for II, III, and I was 1.37, 1.53, and 1.88, respectively.5 Although the IC50 values of II and III were similar, III had more inhibitory effect at high dosages since III was capable of suppressing the T-currents to around 0 at 3 µM (Figure 2(d)), while II was not. Hence, III might be more useful than II in pain therapy because the T-currents were linked to acute peripheral nociception and neuropathic pain in animal models.14,15
In Cav3.1-HEK cells, I, II, and III did not change voltage-dependent activation and steady-state inactivation curves. On the other hand, all 3 PFVNs tended to shift the activation curves toward positive potentials, and II and III, but not I, tended to shift the steady-state inactivation curves toward negative potentials in Cav3.2-HEK cells.5 In these cells, the inhibitory effect of I (SG) on T-currents was irreversible, whereas those of II and III was reversible (Figure 3(a)-(c)). Interestingly, the T-current-blocking effect of II, but not that of I, was reversible (Figure 3(a) and (b)), although both 8-PFVNs had the alkyl side-chains at 8-position (Figure 1(a) and (b)). Hence, the structural characteristic, responsible for the irreversibility of the T-channel blockade of I, is still open to question. However, these results also evoked the supposition that I (SG) with a more lipophilic lavandulyl side-chain (Figure 1(a)-(c)) was able to penetrate into the plasma membrane, which prevented I from washing. Unfortunately, this supposition has not yet been confirmed.
The recovery of T-currents in Cav3.2-HEK cells after treating with prenylflavanones for 5 to 6 minutes, shown as black bar. The inhibitory effect of SG (I) (a) was irreversible, but that of (2S)-8-PNG (II) (b) and (2S)-6-PNG (III) (c) was washable.
Further, (2R/S)-6-PNG (IV), cheaper to synthesize, was compared to III. The result reveals that both III and IV potently inhibited T-currents in Cav3.2-HEK cells, with similar IC50 values, while they exhibited relatively less inhibitory effects on HVA- and Nav-currents in differentiated NG108-15 cells, and the T-channel selectivity of IV was higher than that of III (Table 1).5 Therefore, it is likely that (2R)-6-PNG, if available, might be more selective to T-channel than III, which has yet to be ascertained. Nonetheless, the blockade of III and IV on HVA-calcium channels and Nav-channels might contribute to their analgesic activities, particularly in neuropathic pain models. Indeed, by acting on the α2δ subunit of HVA-calcium and Nav-channels, gabapentinoids are effective for the treatment of neuropathic pain.16,17III and IV are equipotent in the Cav3.2-channel blockade. This means that III and (2R)-6-PNG are also equipotent although the 4′-OH groups on 2 optical isomers are in opposite orientation. This result may lead to conclude inaccurately that the optical configuration of 6-PFVNs is not important for their Cav3.2-channel blockade. Fortunately, it is realized that the 4′-OH groups on phenyl ring of III or (2R)-6-PNG were still inappropriate to the T-channel-blocking action of 6-PNG. This assumption was confirmed by a SAR study showing that the 2′-OH groups, but not the 4′-OH groups, were optimal functional groups for the T-channel-blockade of 6-PNG.
The IC50 Values of (2S)-6-PNG (III) and (2 R/S)-6-PNG (IV) for T-Current Inhibition in Cav3.2-HEK Cells, and HVA- and NaV-Current Inhibition in Neuron-Like Differentiated NG108-15 Cells.5
Compds
IC50 (µM)
T-current
HVA-current
NaV-current
(2S)-6-PNG (III)
0.917
2.005
1.661
(2R/S)-6-PNG (IV)
0.928
4.765
3.250
PNG, prenylnaringenin; HVA, high voltage activated.
In contrast, (2S)-NG (V) and (2R/S)-NG (VI) (Figure 1(e) and (f)) even at 30 µM cannot affect the T-currents (Figure 2(e)), which suggests the importance of alkyl side-chains at 6- or 8-position for T-channel blockade of PFVNs. This assumption was also confirmed by a further SAR study, gone in detail hereinafter.
Assessment of PFVNs on Alleviating Pain in Animal Models
This step employed some animal models to evaluate the efficacy of novel PFVN T-channel blockers on alleviating pain. First, the intraplantar (i.pl.) administration of H2S donors, such as NaHS or Na2S, induces Cav3.2-dependent mechanical hyperalgesia and/or allodynia in rats or mice.9,18,19 These animal models are convenient and useful to evaluate the analgesic effects of T-channel blockers in vivo.10 In our experiments, the mechanical nociceptive threshold of mice decreased maximally at 15 minutes after the i.pl. injection of NaHS at 0.1 nmol/paw (Figure 4(a)-(f)),5 an effect known to be dependent on both Cav3.2 and TRPA1 channels.10,19 This NaHS-induced allodynia in mice was prevented by oral or intraperitoneal (IP) preadministration of the SR-extract at 200 to 400 mg/kg (Figure 4(a) and (b)). Moreover, SG (I), (2S)-8-PNG (II), and (2S)-6-PNG (III), but not (2R/S)-NG (VI), abolished the NaHS-induced allodynia in mice, when such PFVNs were co-administered at 1 or 10 pmol/paw with the i.pl. administration of NaHS (Figure 4(c)-(f)). Hence, the novel PFVN T-channel blockers such as I, II, and III might be useful for pain therapy.
The pharmacological properties of screening prenylflavanones assessed by the von Frey test. (a-f) Effects of SR-extract, SG (I), (2S)-8-PNG (II), (2S)-6-PNG (III), and (2R/S)-naringenin (VI or (2R/S)-NG) on the somatic mechanical allodynia induced by intraplantar (i.pl.) injection of NaHS, an H2S donor, in mice. SR-extract was administered orally or IP at 20 minutes before i.pl. injection of NaHS (a, b). I, II, III, and VI were co-administered i.pl. with NaHS (c-f). Nociceptive thresholds were examined at 15 minutes after i.pl. injection of NaHS or vehicle (V). (g, h) Effect of (2R/S)-6-PNG (IV) on the visceral nociception caused by intracolonic (i.col.) administration of Na2S, another H2S donor, in mice. IV was administered IP at 15 minutes before i.col. injection of Na2S in conscious mice. Visceral pain-like nociceptive behavior was observed and counted for 30 minutes starting immediately after i.col. injection of Na2S (g), and subsequently, referred hyperalgesia was evaluated by von Frey test (h). (i-l) Effects of i.pl. (i, j) or IP (k, l) administration of IV on the neuropathic allodynia induced by partial sciatic nerve ligation (PSNL) or treatment with oxaliplatin (OHP) in mice. IV was administered to mice on eighth day after PSNL (PSNL-mice) or sham operation (Sham-mice) (i, k) and after a single i.p. administration of OHP at 5 mg/kg (OHP-mice) or vehicle (Control-mice) (j, l). (m, n) Disappearance of the suppressing effect of I or IV on the neuropathic mechanical allodynia induced by PSNL in Cav3.2-knockout (Cav3.2-KO) mice. I or IV was administered IP to wild-type C57BL/6J (WT) mice (m) or Cav3.2-KO mice (n) on eighth day after PSNL. Data show the mean ± SEM for 4 to 9 (a-f), 5 to 7 (g, h), 5 to 12 (i-l), or 5 to 6 (m, n) mice. *P < 0.05, **P < 0.01 vs V + V (a-h), V in Sham-mice (i, k), V in Control-mice (j, l), V in Sham-WT-mice (m), or V in Sham-Cav3.2-KO-mice (n); †P < 0.05, ††P < 0.01 vs V + NaHS (a-f), V + Na2S (g, h), V in PSNL-mice (i, k), V in OHP-mice (j, l), or V in PSNL-WT-mice (m).5 SR-extract, sophorae radix extract; SG, sophoraflavanoneG; PNG, prenylnaringenin.
After recognizing (2R/S)-6-PNG (IV) as a novel PFVN T-channel blockers, several other animal models were applied to evaluate the analgesic effects of IV. The effect of systemic administration of IV was assessed on a luminal H2S-induced colonic pain by intracolonic (i.col.) administration of Na2S at 5 nmol/mouse, to produce visceral pain-like nociceptive behavior in mice (Figure 4(g)), followed by referred hyperalgesia in lower mouse abdomen10,20-22 (Figure 4(h)). IV, preadministered IP in a dose of 10 to 30 mg/kg, reduced this Na2S-induced nociceptive behavior and/or referred hyperalgesia significantly (Figure 4(g) and (h)).5 Further, 2 distinct neuropathic pain models, induced by partial sciatic nerve ligation (PSNL) and by treatment with oxaliplatin (OHP) in mice, were used. As a result, the i.pl. administration of IV in the ranges of 0.01 to 1 and 0.1 to 10 nmol/paw restored the mechanical allodynia induced by PSNL (Figure 4(i)) and by the IP administration of OHP (Figure 4(j)), respectively.5 Likewise, the IP administration of IV in the dose of 20 to 30 and 10 to 20 mg/kg significantly reversed the PSNL-induced allodynia (Figure 4(k)) and the OHP-induced allodynia in mice (Figure 4(k)), respectively.5 Therefore, IV is an effective analgesic agent. Taken together, the new PFVN T-channel blockers including I, II, III, and IV are likely to relieve the pain involving the Cav3.2 channel.
In further studies, the novel PFVN T-channel blockers, such as IV and I, have been shown to mediate analgesia through the blockade of the CaV3.2 T-channel in vivo. Indeed, the IP administration of IV or I at 30 mg/kg clearly restored the PSNL-induced allodynia in wild-type C57BL/6J mice (Figure 4(m)), but the anti-allodynic effect of IV and I completely disappeared in the Cav3.2-knockout mice (Figure 4(n)).5
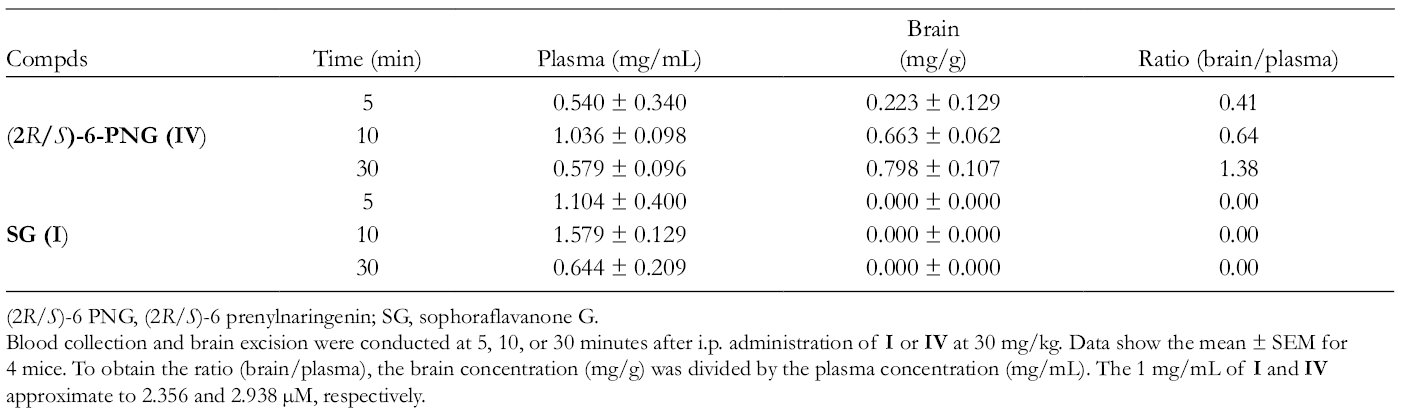
In addition, the plasma and brain tissue levels of IV and I were monitored after their systemic administration. The levels of IV peaked at 10 minutes in plasma and 30 minutes or later in brain after its IP administration at 30 mg/kg (Table 2). The results showed that the levels of IV in plasma and brain were consistent to its Cav3.2 IC50 value in vitro, and IV highly permeates across the blood-brain barrier (BBB) (Tables 1–2). Therefore, the analgesic effect of IV on the neuropathic and colonic pain/referred hyperalgesia (Figure 4(g)-(l)) may be the outcome of the blockade of both peripheral and central CaV3.2 T-channels. Otherwise, I was not detectable in brain for 5 to 30 minutes, although the plasma level of I reached a maximum at 10 minutes after its IP administration at 30 mg/kg (Table 2).5 Hence, the anti-nociceptive activity of I is attributable to the peripheral mechanisms because I does not appear to penetrate into the central nervous system (CNS). In general, the highly BBB-permeable T-channel blockers could be more effective in humans,23,24 but they could cause remarkable effect of sedation which needs to be assessed.5
Time-Related Concentrations of (2R/S)-6-PNG and SG in the Plasma and Brain After Their IP Administration in Mice.5
Compds
Time (min)
Plasma (mg/mL)
Brain(mg/g)
Ratio (brain/plasma)
5
0.540 ± 0.340
0.223 ± 0.129
0.41
(2R/S)-6-PNG (IV)
10
1.036 ± 0.098
0.663 ± 0.062
0.64
30
0.579 ± 0.096
0.798 ± 0.107
1.38
5
1.104 ± 0.400
0.000 ± 0.000
0.00
SG (I)
10
1.579 ± 0.129
0.000 ± 0.000
0.00
30
0.644 ± 0.209
0.000 ± 0.000
0.00
(2R/S)-6 PNG, (2R/S)-6 prenylnaringenin; SG, sophoraflavanone G.
Blood collection and brain excision were conducted at 5, 10, or 30 minutes after i.p. administration of I or IV at 30 mg/kg. Data show the mean ± SEM for 4 mice. To obtain the ratio (brain/plasma), the brain concentration (mg/g) was divided by the plasma concentration (mg/mL). The 1 mg/mL of I and IV approximate to 2.356 and 2.938 µM, respectively.
The cardiovascular and central side effects were also evaluated. On the endothelium-removed rat aortic ring precontracted with 50 mM KCl solution, I, III, and IV up to 30 µM caused a slight relaxation, but II in levels of 10 to 30 µM greatly induced relaxation.5 Further, II is well known as a potent hop-derived estrogenic compound,25 and its agonistic activity on estrogen receptors alters according to varying lengths and branching patterns of the alkyl side-chain at 8-position,26 while III does not have such estrogenic activity.27 Next, the IP administration of IV at 20 mg/kg only slightly affected mean blood pressure and had no effect on heart rate in conscious mice.5 Finally, IV, administrated IP at 30 mg/kg, did not cause any significant changes in the open-field behavior or in rota-rod performance of mice.5
Recently, a number of T-channel blockers have been developed and evaluated for anti-hyperalgesic or anti-allodynic activity in distinct pain models.28 The IC50 values for T-current blocking (around 1 µM) and the IP anti-hyperalgesic/allodynic doses (10, 30 mg/kg) of IV or III are similar to those of many good T-channel blockers such as NNC 55-0396 and RQ-00311651,10,12,29 except TTA-A2 with the IC50 value for T-channel inhibition less than 0.1 µM, and the IP anti-hyperalgesic/allodynic dose around 1 mg/kg.30,31 However, it is noteworthy that TTA-A2 easily penetrates into the CNS and causes suppression of active wake and promotion of slow-wave sleep.31 Thus, III and IV could be considered as analgesics with a well-balanced BBB permeability.
Synthesis of 6-PFVNs for SAR Studies
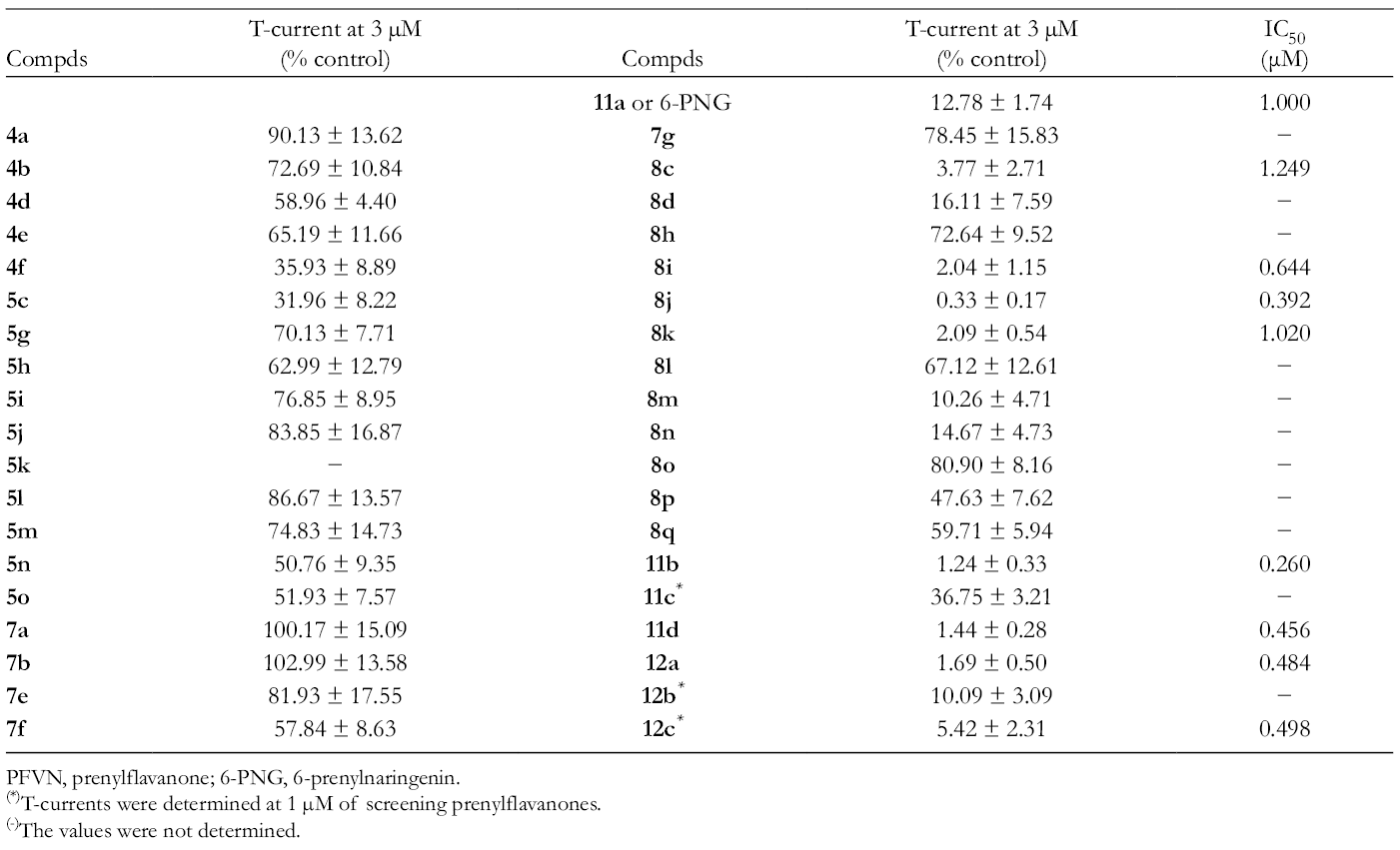
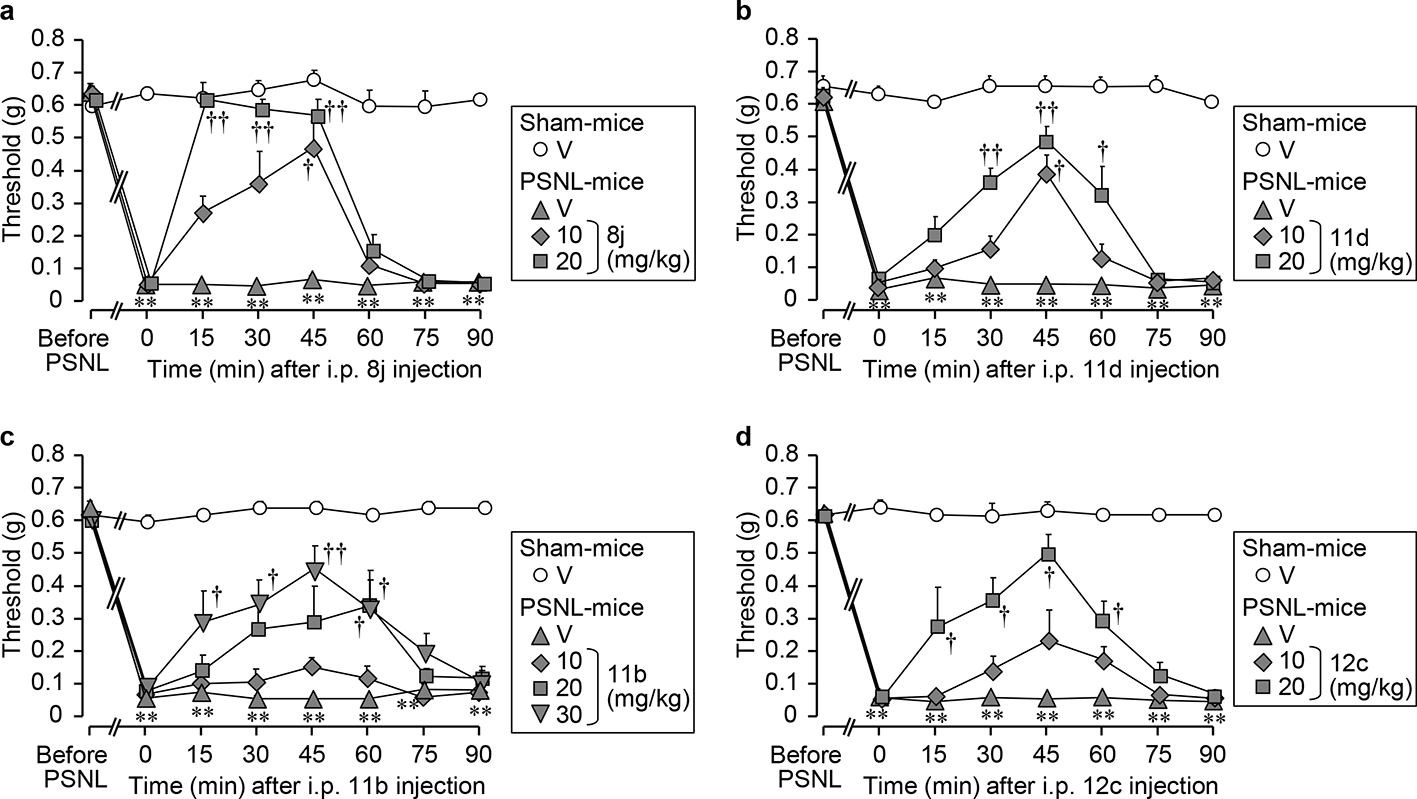
The SAR studies, using the synthetic 6-PFVNs, are highlighted in Schemes 1 and 2. As mentioned above, preliminary SAR consideration based on I, II, III, and IV suggested that III and IV were the most useful for pain therapy. Thus, further SAR studies will focus on 6-PFVN derivatives of IV (Figure 5). The prenyl group at 6-position might play an important role in the T-channel blockade of 6-PNG, so the first thing was to synthesize the 6-PNG derivatives with the 6-allyl-substituent, varied in the length of carbon chain and the degree of saturation, according to Scheme 2.6 As a result, the 6-PNG derivatives with the slightly large 6-allyl-substituent including 11b and 11d, or a little smaller 6-saturated substituent including 12a and 12 c, showed the potent T-channel blocking effects (Scheme 2). In contrast, 11c with a 3,3-di-n-propylallyl (DPA) group at 6-position exhibited a less potent T-channel suppression in vitro (Scheme 2). This result might be caused by the 6-DPA group that hindered the hydroxy groups at 5- or 7-position from reaching their binding sites. In comparison of 11c to 11b, the 5- or 7-hydroxy groups on aromatic ring may also play an important role in the blocking action of 6-PNG. To confirm this assumption, the 6-PFVN derivatives with/without a 7-OH group were synthesized according to Scheme 1.6 Consequently, the crucial role of this 7-OH group in the T-channel blockage of 6-PNG was realized when comparing 8i with 8c, and 8j with 8d (Scheme 1). Moreover, based on the assumption that deals with the inappropriateness of the 4′-OH groups, as mentioned above, the 6-PFVN derivatives with hydroxy groups on the phenyl ring, altered in their positions and quantities, were synthesized according to Scheme 1.6 As a result, the 2′-OH group is able to increase the T-channel blockade of 6-PFVNs because 8i is about 2 times more potent than 8k and 11a (IV or 6-PNG) (Schemes 1, 2 and Table 3). On the other hand, 8i also suppresses the T-current better than 8l, 8m, 8n, and 8o which possess 2 hydroxy groups on the phenyl ring at 2 distinct positions (Scheme 1 and Table 3). According to the SAR studies, we selected 4 candidates (11b, 11d, 12c, and 8j) for the in vivo anti-allodynic activity in a mouse neuropathic pain model.
Reagents and conditions: (a) (Me)2C=CH2 or (Et)2C=CH2 or (n-Pr)2C=CH2, Grubbs catalyst, second generation, benzene, 100°C, sealed tube; (b) K2CO3, MeOH, rt; (c) H2, Pd/C, EtOAc, rt. Tcur (1 µM) is T-current (percentage control) determined at 1 µM of screening PFVNs.
Designation of screening 6-PNG derivatives.
T-Current Suppression of the Synthetic Derivatives in Human CaV3.2-HEK293 Cells and the IC50 Values for T-Current Inhibition of Potent Action Compounds.6
(*)T-currents were determined at 1 µM of screening prenylflavanones.
(-)The values were not determined.
The 4 selected derivatives significantly alleviated the PSNL-induced allodynia in mice, as shown in Figure 6(a)-(d).6 Among them, 11b exhibited an anti-allodynic effect in the doses of 10 to 30 mg/kg, similar to 11a (IV or 6-PNG), while 11d, 12c, and 8j showed the anti-allodynic effects in the doses of 10 to 20 mg/kg, better than 11a (6-PNG) (Figures 4(k) and 6(a)-(d)). Particularly, 8j showed most potent anti-allodynic effect. The anti-allodynic effect of 10 mg/kg of 8j, suppressing the PSNL-induced allodynia in mice, equals the anti-allodynic effect of 30 mg/kg of 11a (6-PNG) (Figures 4(k) and 6(a)).
In the in vivo evaluations of 4 selected derivatives (a) 8j, (b) 11d, and (d) 12c at 10 or 20 mg/kg, and (c) 11b at 10, 20, or 30 mg/kg, or vehicle (V; 0.5% carboxymethyl cellulose) were administered IP to mice 7 days after partial sciatic nerve ligation (PSNL). Nociceptive threshold in the ipsilateral hind paw was assessed by the von Frey test. Data show the mean with SEM for 5 to 7 mice. **P < 0.01 vs vehicle in sham. †P < 0.05, ††P < 0.01 vs vehicle in PSNL.
Prospects and Challenges of Other Prenylated Flavonoids
Although we have recognized 6-PNG and 8j as novel T-channel blockers, the T-channel blockades of other prenylated flavonoids sub-classes have not yet been assessed. This assessment is an immense challenge, but it will provide a more overall look about SAR consideration of the T-channel blockades of prenylated flavonoids, will have the individual predictions about the relationship between the chemical structure of prenylated flavonoids and their T-channel blocking actions. By our efforts and calling the efforts of other research teams, we hope to obtain more information in near future.
Humulus lupulus L. (Hops) is a long used medicinal product25 and a popular botanical dietary supplement used by women as a sleep aid and for postmenopausal symptom relief.32 Among prenylated phenols in hops, 8-PNG is the most potent phytoestrogen and has received more attention than others including xanthohumol (XN), isoxanthohumol and 6-PNG. The pharmacokinetics of 6-PNG in women was shown by van Breemen et al when they investigated the pharmacokinetics of prenylated hop phenols in women following oral administration of a standardized extract of hops. This human study indicated that the half-life of 6-PNG is long (T1/2 over 21 hours), but there is no acute toxicities.33 The long half-life of 6-PNG may be contributed by a metabolic process of XN.32 In our previous study, the safety of 6-PNG was assessed in the cardiovascular and central side effects in animal models.5 Thereby, other prenylated flavonoids, potent T-channel blockers and useful for pain therapy, should also be considered for their pharmacokinetics and safety in human models.
Due to the limited distribution of prenylated flavonoids in nature, chemical synthesis is a natural choice for researchers interested on such compounds.3 6-PNG can be synthesized from commercially available naringenin according to Scheme 2,34 thereby other 6-PNG derivatives with a larger 6-alkyl-substituent can be also synthesized.6 Moreover, 6-PNG derivatives with other flavanone skeletons were synthesized according to Scheme 1.6 Chemical synthesis appears to be rather flexible, so this technique is useful in the preparation of screening prenylated flavonoids for SAR study and clinical research. Biotransformation by metabolic engineering is another technique to synthesize desired prenylated flavonoids. The key of this technique is to find the transferase that is effective for the desired prenylation on a flavonoid skeleton.3 For example 6-prenyl apigenin, 6-prenyl-4′,5,7-trihydroxyflavone, has already been synthesized by biotransformation.35 Biotransformation is a promising technique, but requires more research.
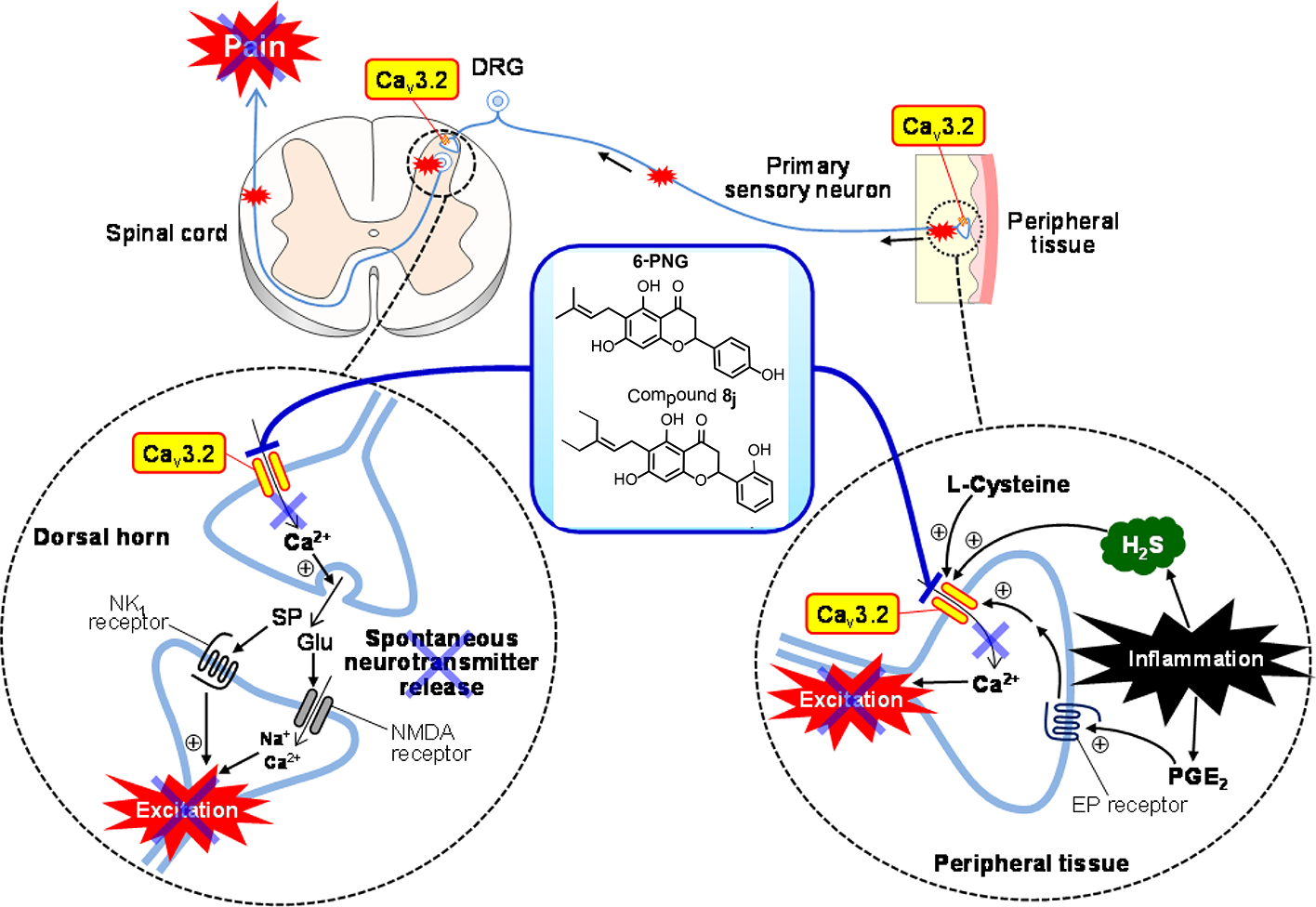
All information about considering PFVNs as the novel T-channel blockers, useful for pain therapy, was offered in this review. III ((2S)-6-PNG) and IV ((2R/S)-6-PNG) are considered as the potent T-channel blockers and alleviate T-channel-dependent somatic and visceral pain in mice without cardiovascular and behavioral side effects. Moreover, a series of 6-PNG derivatives were synthesized. Among them, 8j (KTt45), 6-(3-ethylpent-2-enyl)-5,7-dihydroxy-2-(2-hydroxyphenyl)chroman-4-one, also showed a more potent blockade activity against the human Cav3.2-HEK cells with the IC50value of 0.39 µM, and exhibited a remarkable anti-allodynic effect at 10 mg/kg and achieved almost complete inhibition of the PSNL-induced neuropathy at 20 mg/kg. These compounds are considered to suppress the Cav3.2 T-channels expressed in the peripheral endings and the central terminals of the primary sensory neurons, followed by the attenuation of pain sensation by inhibition of neuronal excitation, and the reduction in spontaneous release of neurotransmitters (Figure 7). The review also called for an effort on SAR consideration of other prenylated flavonoids that may become promising T-channel blockers and also mentioned the challenges of other prenylated flavonoids such as the lack of clinical research, the low abundance in nature, and difficulty in synthesis.
Scheme of mechanisms for the anti-nociceptive effects of 6-PNG and compound 8j. Cav3.2, Cav3.2 T-type Ca2+ channels; SP, substance P; Glu, glutamiate; H2S, hydrogen sulfide.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2014, 2018) (S1411037) and the “Antiaging” Project for Private Universities.
BottaB.VitaliA.MenendezP.MisitiD.MonacheG. Prenylated flavonoids: pharmacology and biotechnology. Curr Med Chem. 2005;12(6):713-739.doi:10.2174/0929867053202241
5.
SekiguchiF.FujitaT.DeguchiTet al. Blockade of T-type calcium channels by 6-prenylnaringenin, a hop component, alleviates neuropathic and visceral pain in mice. Neuropharmacology. 2018;138:232-244.doi:10.1016/j.neuropharm.2018.06.020
6.
Du NguyenH.OkadaT.KitamuraSet al. Design and synthesis of novel anti-hyperalgesic agents based on 6-prenylnaringenin as the T-type calcium channel blockers. Bioorg Med Chem. 2018;26(15):4410-4427.doi:10.1016/j.bmc.2018.07.023
7.
TodorovicSM.Jevtovic-TodorovicV. T-Type voltage-gated calcium channels as targets for the development of novel pain therapies. Br J Pharmacol. 2011;163(3):484-495.doi:10.1111/j.1476-5381.2011.01256.x
8.
KawabataA.IshikiT.NagasawaKet al. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132(1-2):74-81.doi:10.1016/j.pain.2007.01.026
9.
SekiguchiF.KawabataA. T-Type calcium channels: functional regulation and implication in pain signaling. J Pharmacol Sci. 2013;122(4):244-250.doi:10.1254/jphs.13R05CP
10.
SekiguchiF.KawaraY.TsubotaMet al. Therapeutic potential of RQ-00311651, a novel T-type Ca2+ channel blocker, in distinct rodent models for neuropathic and visceral pain. Pain. 2016;157(8):1655-1665.doi:10.1097/j.pain.0000000000000565
11.
TakahashiT.AokiY.OkuboKet al. Upregulation of Ca(v)3.2 T-type calcium channels targeted by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Pain. 2010;150(1):183-191.doi:10.1016/j.pain.2010.04.022
12.
MatsunamiM.MikiT.NishiuraKet al. Involvement of the endogenous hydrogen sulfide/Ca 3.2 T-type Ca 2+ channel pathway in cystitis-related bladder pain in mice. Br J Pharmacol. 2012;167(4):917-928.doi:10.1111/j.1476-5381.2012.02060.x
13.
ZamponiGW.StriessnigJ.KoschakA.DolphinAC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67(4):821-870.doi:10.1124/pr.114.009654
14.
ChoiK-H. The design and discovery of T-type calcium channel inhibitors for the treatment of central nervous system disorders. Expert Opin Drug Discov. 2013;8(8):919-931.doi:10.1517/17460441.2013.796926
15.
LeeJ-H.SeoSH.LimEJet al. Synthesis and biological evaluation of 1-(isoxazol-5-ylmethylaminoethyl)-4-phenyl tetrahydropyridine and piperidine derivatives as potent T-type calcium channel blockers with antinociceptive effect in a neuropathic pain model. Eur J Med Chem. 2014;74:246-257.doi:10.1016/j.ejmech.2013.12.056
16.
BouhassiraD.AttalN. Translational neuropathic pain research: a clinical perspective. Neuroscience. 2016;338:27-35.doi:10.1016/j.neuroscience.2016.03.029
17.
CardosoFC.LewisRJ. Sodium channels and pain: from toxins to therapies. Br J Pharmacol. 2018;175(12):2138-2157.doi:10.1111/bph.13962
18.
MaedaY.AokiY.SekiguchiFet al. Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of Cav3.2 T-type calcium channels. Pain. 2009;142(1-2):127-132.doi:10.1016/j.pain.2008.12.021
19.
OkuboK.MatsumuraM.KawaishiYet al. Hydrogen sulfide-induced mechanical hyperalgesia and allodynia require activation of both Cav3.2 and TRPA1 channels in mice. Br J Pharmacol. 2012;166(5):1738-1743.doi:10.1111/j.1476-5381.2012.01886.x
20.
MatsunamiM.TaruiT.MitaniKet al. Luminal hydrogen sulfide plays a pronociceptive role in mouse colon. Gut. 2009;58(6):751-761.doi:10.1136/gut.2007.144543
21.
MatsunamiM.KirishiS.OkuiT.KawabataA. Chelating luminal zinc mimics hydrogen sulfide-evoked colonic pain in mice: possible involvement of T-type calcium channels. Neuroscience. 2011;181:257-264.doi:10.1016/j.neuroscience.2011.02.044
22.
Tsubota-MatsunamiM.NoguchiY.OkawaY.SekiguchiF.KawabataA. Colonic hydrogen sulfide-induced visceral pain and referred hyperalgesia involve activation of both Ca(v)3.2 and TRPA1 channels in mice. J Pharmacol Sci. 2012;119(3):293-296.doi:10.1254/jphs.12086SC
23.
SerraJ.DuanWR.LockeC.SolàR.LiuW.NothaftW. Effects of a T-type calcium channel blocker, ABT-639, on spontaneous activity in C-nociceptors in patients with painful diabetic neuropathy: a randomized controlled trial. Pain. 2015;156(11):2175-2183.doi:10.1097/j.pain.0000000000000249
24.
ZieglerD.DuanWR.AnG.ThomasJW.NothaftW. A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain. 2015;156(10):2013-2020.doi:10.1097/j.pain.0000000000000263
25.
KeilerAM.ZierauO.KretzschmarG. Hop extracts and hop substances in treatment of menopausal complaints. Planta Med. 2013;79(7):576-579.doi:10.1055/s-0032-1328330
26.
RoelensF.HeldringN.DhoogeWet al. Subtle side-chain modifications of the hop phytoestrogen 8-prenylnaringenin result in distinct agonist/antagonist activity profiles for estrogen receptors alpha and beta. J Med Chem. 2006;49(25):7357-7365.doi:10.1021/jm060692n
27.
OverkCR.YaoP.ChadwickLRet al. Comparison of the in vitro estrogenic activities of compounds from hops (Humulus lupulus) and red clover (Trifolium pratense). J Agric Food Chem. 2005;53(16):6246-6253.doi:10.1021/jf050448p
28.
SnutchTP.ZamponiGW. Recent advances in the development of T-type calcium channel blockers for pain intervention. Br J Pharmacol. 2018;175(12):2375-2383.doi:10.1111/bph.13906
29.
OkuboK.TakahashiT.SekiguchiFet al. Inhibition of T-type calcium channels and hydrogen sulfide-forming enzyme reverses paclitaxel-evoked neuropathic hyperalgesia in rats. Neuroscience. 2011;188:148-156.doi:10.1016/j.neuroscience.2011.05.004
30.
FrancoisA.KerckhoveN.MeleineMet al. State-dependent properties of a new T-type calcium channel blocker enhance Ca(V)3.2 selectivity and support analgesic effects. Pain. 2013;154(2):283-293.doi:10.1016/j.pain.2012.10.023
31.
KrausRL.LiY.GreganYet al. In vitro characterization of T-type calcium channel antagonist TTA-A2 and in vivo effects on arousal in mice. J Pharmacol Exp Ther. 2010;335(2):409-417.doi:10.1124/jpet.110.171058
32.
WangS.DunlapTL.HowellCEet al. Hop (Humulus lupulus L.) extract and 6-Prenylnaringenin induce P450 1A1 catalyzed estrogen 2-hydroxylation. Chem Res Toxicol. 2016;29(7):1142-1150.doi:10.1021/acs.chemrestox.6b00112
33.
van BreemenRB.YuanY.BanuvarSet al. Pharmacokinetics of prenylated hop phenols in women following oral administration of a standardized extract of hops. Mol Nutr Food Res. 2014;58(10):1962-1969.doi:10.1002/mnfr.201400245
34.
TischerS.MetzP. Selective C-6 Prenylation of Flavonoidsvia Europium(III)- Catalyzed Claisen Rearrangement and Cross-Metathesis. Adv Synth Catal. 2007;349(1-2):147-151.doi:10.1002/adsc.200600454
35.
YangX.YangJ.JiangYet al. Regiospecific synthesis of prenylated flavonoids by a prenyltransferase cloned from Fusarium oxysporum. Sci Rep. 2016;6(1):1-10.doi:10.1038/srep24819