
Abstract
Five new triterpene glycosides, psolusosides C3 (
The investigation of a complicated glycosidic sum of the sea cucumber Psolus fabricii (Psolidae, Dendrochirotida) was started so long ago as in 1982, but many glycosides at the time could not be isolated as pure compounds. Recently we have decided to make a new attempt using modern technical base. This attempt has resulted in the isolation of 3 new hexaosides, psolusosides C1, C2, and D1 as well as 5 known compounds.
1
Herein, we report the isolation and structural elucidation of 5 additional hexaosides, named as psolusosides C3 (

Structures of the glycosides: 1—psolusoside C3; 2—psolusoside D2; 3—psolusoside D3; 4—psolusoside D4; 5—psolusoside D5.
The initial stages of isolation of the fractions, containing the psolusosides of the groups C and D, were reported earlier.
1
Subsequently these fractions were subjected to reversed-phase high-performance liquid chromatography (HPLC) to give individual psolusosides C3 (
The 1H and 13C NMR spectra of carbohydrate part of psolusoside C3 (
13C and 1 H Nuclear Magnetic Resonance Chemical Shifts, HMBC and ROESY Correlations of the Carbohydrate Moiety of Psolusoside C3 (1).
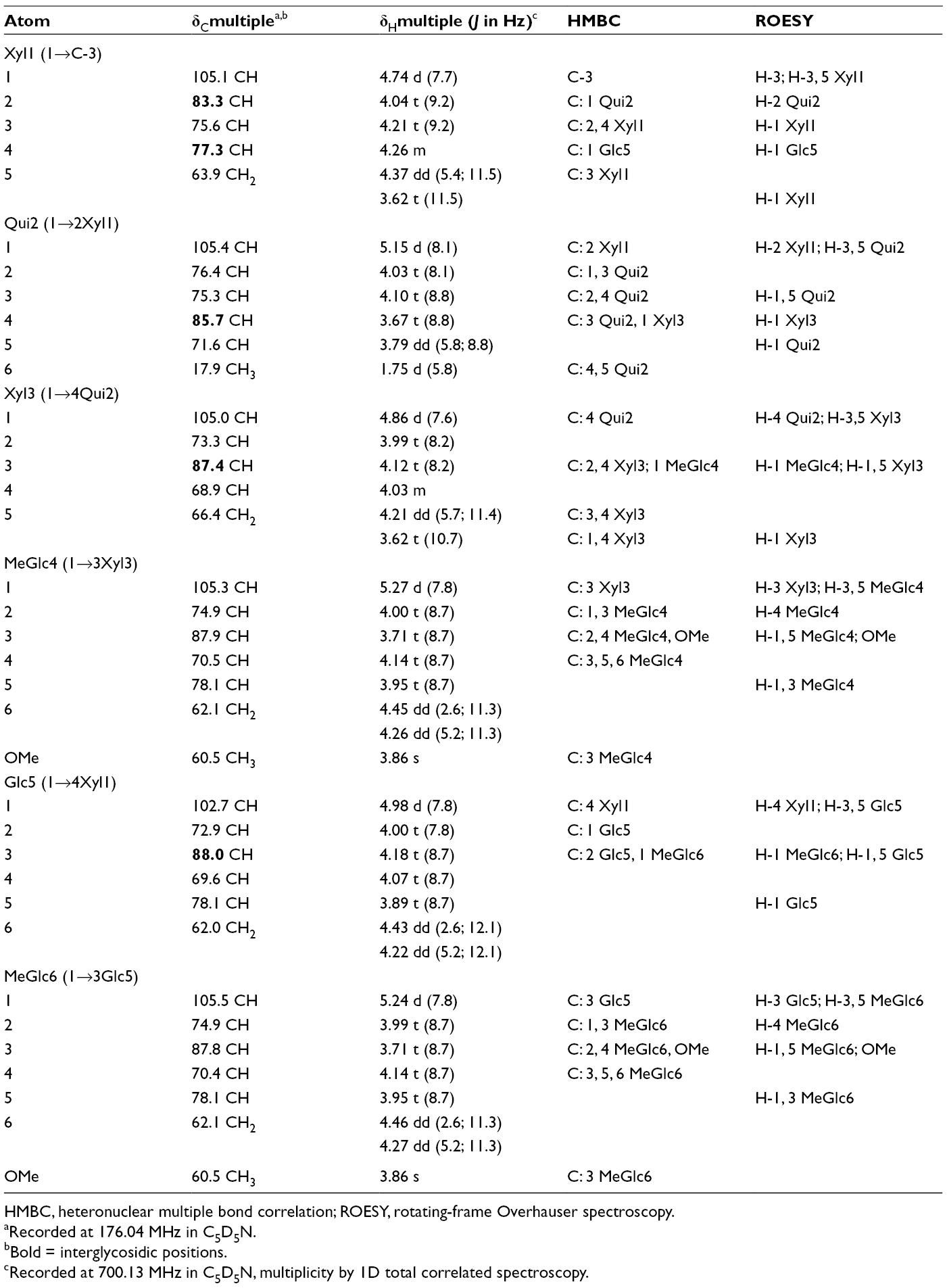
HMBC, heteronuclear multiple bond correlation; ROESY, rotating-frame Overhauser spectroscopy.
aRecorded at 176.04 MHz in C5D5N.
bBold = interglycosidic positions.
cRecorded at 700.13 MHz in C5D5N, multiplicity by 1D total correlated spectroscopy.
Analysis of the 1H and 13C NMR spectra (Table 2) of the aglycone moiety of psolusoside C3 (
13C and 1H Nuclear Magnetic Resonance Chemical Shifts, HMBC and ROESY Correlations of Aglycone Moiety of Psolusosides C3 (1) and D5 (5).

HMBC, heteronuclear multiple bond correlation; ROESY, rotating-frame Overhauser spectroscopy.
aRecorded at 176.04 MHz in C5D5N.
bRecorded at 700.13 MHz in C5D5N, multiplicity by one-dimensional total correlated spectroscopy.
The same side chain frequently occurred in the glycosides of sea cucumbers belonging to different taxa.
4
However, its combination with 16-ketoholost-9(11)-ene polycyclic system has never been found earlier. Thus, the aglycone of psolusoside C3 (
All these data indicated that psolusoside C3 (
The 1H and 13C NMR spectra of carbohydrate moieties of psolusosides D2 to D5 (
13C and 1H Nuclear Magnetic Resonance Chemical Shifts, HMBC and ROESY Correlations of Carbohydrate Moiety of Psolusosides D2 to D5 (
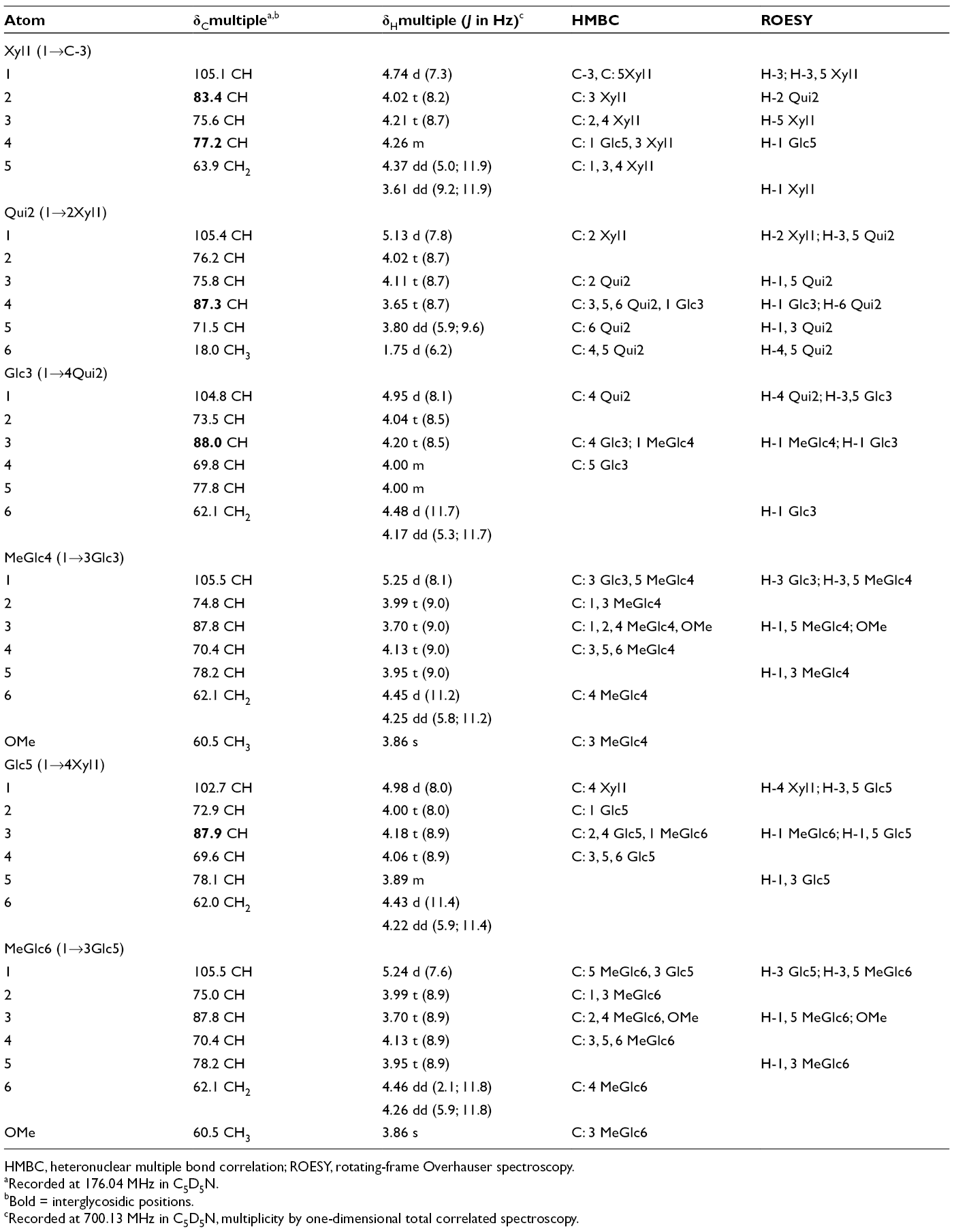
HMBC, heteronuclear multiple bond correlation; ROESY, rotating-frame Overhauser spectroscopy.
aRecorded at 176.04 MHz in C5D5N.
bBold = interglycosidic positions.
cRecorded at 700.13 MHz in C5D5N, multiplicity by one-dimensional total correlated spectroscopy.
The presence of the signals assigned to methine (δC 77.8) and hydroxymethyl groups (δC 62.1) characteristic of C-5 and C-6 of glucose residue positioned this glucose as the third sugar unit in psolusosides D2 to D5 (
The 1H and 13C NMR spectra of the aglycone part of psolusoside D2 (
13C and 1H Nuclear Magnetic Resonance Chemical Shifts, HMBC and ROESY Correlations of Aglycone Moiety of Psolusoside D2 (2).

ROESY, rotating-frame Overhauser spectroscopy; HMBC, heteronuclear multiple bond correlation.
aRecorded at 176.04 MHz in C5D5N.
bRecorded at 700.13 MHz in C5D5N, multiplicity by one-dimensional total correlated spectroscopy.
The molecular formula of psolusoside D2 (
Thus, psolusoside D2 (
The signals in the 1H and 13C NMR spectra of the aglycone part of psolusoside D3 (
13C and 1H Nuclear Magnetic Resonance Chemical Shifts, HMBC and ROESY Correlations of Aglycone Moiety of Psolusoside D3 (3).

HMBC, heteronuclear multiple bond correlation; ROESY, rotating-frame Overhauser spectroscopy.
aRecorded at 176.04 MHz in C5D5N.
bRecorded at 700.13 MHz in C5D5N, multiplicity by one-dimensional total correlated spectroscopy.
The finding of hydroperoxy groups in the triterpene glycosides from sea cucumbers is very unusual. But recently the glycoside with the same side chain as psolusoside D3 (
The fragmentary ions in the (−) ESI MS/MS of ion [M − H]− at m/z 1453.6 (see Experimental) confirmed the sequence of monosaccharide units in the sugar chain of
Thus, psolusoside D3 (
The signals in the 1H and 13C NMR spectra of the aglycone part of psolusoside D4 (
13C and 1H Nuclear Magnetic Resonance Chemical Shifts, HMBC and ROESY Correlations of Aglycone Moiety of Psolusoside D4 (4).

HMBC, heteronuclear multiple bond correlation; ROESY, rotating-frame Overhauser spectroscopy.
aRecorded at 176.04 MHz in C5D5N.
bRecorded at 700.13 MHz in C5D5N, multiplicity by one-dimensional total correlated spectroscopy.
The molecular formula of psolusoside D4 (
Thus, psolusoside D4 (
The aglycone of psolusoside D5 (
The molecular formula of psolusoside D5 (
Thus, psolusoside D5 (
It is interesting to note that the series of the side chains characteristic of psolusosides C1
1
and D4 (
The cytotoxic activities of compounds
Hemolytic Activities of Glycosides 1 to

Psolusosides C1 and C3 (
Experimental
General Experimental Procedures
Specific rotation, Perkin-Elmer 343 Polarimeter; NMR, AVANCE III-700 Bruker spectrometer at 700.13 MHz/176.04 MHz (1H/13C); ESI MS (positive ion mode), Agilent 6510 Q-TOF apparatus, sample concentration 0.01 mg/mL; HPLC, Agilent 1100 apparatus with a differential refractometer; column Supelco Ascentis RP-Amide (10 × 250 mm, 5 µm).
Animal Material
Specimens of the sea cucumber Psolus fabricii (family Psolidae; order Dendrochirotida) were collected in the Sea of Okhotsk near Onekotan Island (Kurile Islands). Sampling was performed with a scallop dredge in August-September, 1982, at a depth of 100 m during expeditional works on fishing seiners “Mekhanik Zhukov” and “Dalarik.” Sea cucumbers were identified by Prof V.S. Levin; voucher specimens are preserved in A.V. Zhirmunsky National Scientific Center of Marine Biology, Vladivostok, Russia.
Extraction and Isolation
The sea cucumbers were extracted twice with refluxing 60% EtOH. The extract was evaporated to water residuum and lyophilized followed by the extraction with CHCl3/MeOH (1:1). The obtained extract was evaporated and submitted to the subsequent extraction by EtOAc/H2O to remove the lipid fraction. The water layer remaining after this extraction was chromatographed on a Polychrom-1 column (powdered Teflon, Biolar, Latvia). The glycosides were eluted with 50% EtOH, evaporated, and subsequently chromatographed on Si gel column with CHCl3/EtOH mixtures in ratios 10:1 and 5:1 as mobile phase, followed by the gradient CHCl3/EtOH/H2O (100:50:4), (100:75:10), (100:100:17), (100:150:50) as mobile phase to give several subfractions containing different groups of glycosides. The subfractions were kept at the temperature -18°C. The fractions 1 (205 mg) and 2 (305 mg) were obtained after additional rechromatography on Si gel columns of the most nonpolar subfractions with CHCl3/EtOH/H2O (100:50:4) and (100:75:10), correspondingly, as mobile phase. Fraction 1 was submitted to HPLC on Supelco Ascentis RP-Amide column with 45% acetonitrile (MeCN) as mobile phase to give subfractions 1 to 6. The rechromatography of subfraction 1 in 30% MeCN gave 2.5 mg of psolusoside C3 (
Psolusoside C3 (1)
Colorless powder.
[α]D 20: –20 (c 0.1, 50% MeOH).
HR ESI MS (−) m/z: 1407.6404 (calculated 1407.6438) [M − H]−; HR ESI MS (+) m/z: 1431.6347 (calculated 1431.6403) [M + Na]+, 727.3129 (calculated 727.3148) [M + 2Na]2+; ESI MS/MS (+) m/z: 1255.6 [M + Na – MeGlc +H]+, 1123.5 [M + Na – MeGlc – Xyl +H]+, 1093.6 [M + Na – MeGlc – Glc +H]+, 977.5 [M + Na – MeGlc – Xyl – Qui +H]+, 965.3 [M + Na – C30H43O4(Agl) +H]+, 947.4 [M + Na – 2MeGlc – Xyl +2 H]+,789.3 [M + Na – C30H43O4– MeGlc +2 H]+, 657.2 [M + Na – C30H43O4 – MeGlc – Xyl +2 H]+, 511.2 [M + Na – C30H43O4 – MeGlc – Xyl – Qui +2 H]+.
Psolusoside D2 (2)
Colorless powder.
[α]D 20: –45 (c 0.1, 50% MeOH).
NMR: See Tables 3 and 4. HR ESI MS (+) m/z:1459.6310 (calculated 1459.6352) [M + Na]+; 741.3105 (calculated 741.3122) [M + 2Na]2+; ESI MS/MS (+) m/z: 1283.6 [M + Na – MeGlc +H]+, 1121.5 [M + Na – MeGlc – Glc +H]+, 995.3 [M + Na – C30H41O4 (Agl) +H]+, 975.4 [M + Na – MeGlc – Glc – Qui +H]+, 945.4 4 [M + Na – 2MeGlc – Glc +2 H]+, 819.3 [M + Na – C30H41O4 – MeGlc +2 H]+, 799.4 4 [M + Na – 2MeGlc – Glc – Qui +2 H]+, 657.2[M + Na – C30H41O4 – MeGlc – Glc +2 H]+.
Psolusoside D3 (3)
Colorless powder.
[α]D 20: –77 (c 0.1, 50% MeOH).
NMR: See Tables 3 and 5. HR ESI MS (−) m/z: 1453.6442 (calculated 1453.6493 for C67H105O34) [M − H]−; ESI MS/MS (−) m/z:1377.6 [M − H – C3H7O2 + H]−, 1277.6 [M − H – MeGlc +H]−, 1201.6 [M − H – C3H7O2−MeGlc +2 H]−, 1115.5 [M − H – MeGlc – Glc +H]−, 1039.5 [M − H – C3H7O2−MeGlc – Glc +2 H]−, 955.4 [M − H – Agl +H]−, 777.4 [M − H – 2MeGlc – 2Glc +2 H]−.
Psolusoside D4 (4)
Colorless powder.
[α]D 20: –52 (c 0.1, 50% MeOH).
HR ESI MS (+) m/z: 1461.6478 (calculated 1461.6509) [M + Na]+, 742.3192 (calculated 742.3200) [M + 2Na]2+; HR ESI MS (−) m/z: 1437.6526 (calculated 1437.6544) [M – H]−;ESI MS/MS (+) m/z: 1285.6 [M + Na – MeGlc +H]+, 1123.5 [M + Na – MeGlc – Glc +H]+, 995.3 [M + Na – C30H43O4 (Agl) +H]+, 977.4 [M + Na – MeGlc – Glc – Qui +H]+, 947.4 4 [M + Na – 2MeGlc – Glc +2 H]+, 819.3 [M + Na – C30H43O4 – MeGlc +2 H]+, 657.2 [M + Na – C30H43O4 – MeGlc – Glc +2 H]+.
Psolusoside D5 (5)
Colorless powder.
[α]D 20: –73 (c 0.1, 50% MeOH).
HR ESI MS (+) m/z: 1461.6477 (calculated 1461.6509) [M + Na]+; 742.3187 (calculated 742.3200) [M + 2Na]2+.
HR ESI MS (−) m/z: 1437.6519 (calculated 1437.6544) [M – H]−; ESI MS/MS (+) m/z: 1285.6 [M + Na – MeGlc +H]+, 1123.5 [M + Na – MeGlc – Glc +H]+, 995.3 [M + Na – C30H43O4 (Agl) +H]+, 977.4 [M + Na – MeGlc – Glc – Qui +H]+, 947.4 4 [M + Na – 2MeGlc – Glc +2 H]+, 819.3 [M + Na – C30H43O4 – MeGlc +2 H]+, 657.2 [M + Na – C30H43O4 – MeGlc – Glc +2 H]+.
Cell Culture
The museum tetraploid strain of murine ascite Ehrlich carcinoma cells from the All-Russian oncology center (Moscow, Russia) was used. The cells were separated from the ascites, which were collected on day 7 after inoculation in mouse CD-1 line. The cells were washed of the ascites triply and resuspended in RPMI-1640 medium containing 8 µg/mL gentamicin (BioloT, Russia). Neuro 2A cells were cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum (BioloT, Russia) and 1% penicillin/streptomycine (Invitrogen). Cells were incubated at 37°C in humidified atmosphere containing 5% (v/v) CO2.
Cytotoxic Activity
Cytotoxic activities were investigated by Nonspecific Esterase Activity Assay. Ten microliters of the test substance solution and 100 µL of the cell suspension were placed into each well of a 96-well microplate. The plate was incubated in a CO2 incubator at 37°C for 1 or 24 hours. A stock solution of the probe fluorescein diacetate (FDA; Sigma) in dimethyl sulfoxide (1 mg/mL) was prepared. After incubation of the cells with test compounds, 10 µL of FDA solution (50 µg/mL) was added to each well and the plate was incubated at 37°C for 15 minutes. Cells were washed with phosphate-buffered saline (PBS), and fluorescence was measured with a Fluoroskan Ascent plate reader (Thermo Labsystems, Finland) at λex = 485 nm and λem = 518 nm. All experiments were repeated in triplicate. Cytotoxic activity was expressed as the percent of cell viability.
Hemolytic Activity
Blood was taken from CD-1 mice (18–20 g). The mice were anesthetized with diethyl ether, their chests were rapidly opened, and blood was collected in cold (4°C) 10 mM PBS, pH 7.4 without anticoagulant. Erythrocytes were washed 3 times in PBS using at least 10 vol. of washing solution by centrifugation (2 000 rpm) for 5 minutes. Erythrocytes were used at a concentration that provided an optical density of 1.0 at 700 nm for a nonhemolyzed sample. Twenty microliters of a water solution of test substance with a fixed concentration was added to a well of a 96-well plate containing 180 µL of the erythrocyte suspension. Erythrocyte suspension was incubated with substances for 24 hours at 37°C. After that, the optical density of the obtained solutions was measured and ED50 for hemolytic activity of each compound was calculated. 11
Footnotes
Acknowledgments
The study was carried out on the equipment of the Collective Facilities Center “The Far Eastern Center for Structural Molecular Research (NMR/MS) PIBOC FEB RAS”. The authors are also appreciative to Professor Vladimir Koval (Institute of Chemical Biology & Fundamental Medicine, Novosibirsk, Russia) for assistance in mass-spectrometric studies and Professor Valentin A. Stonik (G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Vladivostok, Russia) for reading and discussion of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author(s) received partial financial support from the Grant of the Russian Foundation of Basic Research No. 19-04-000-14.