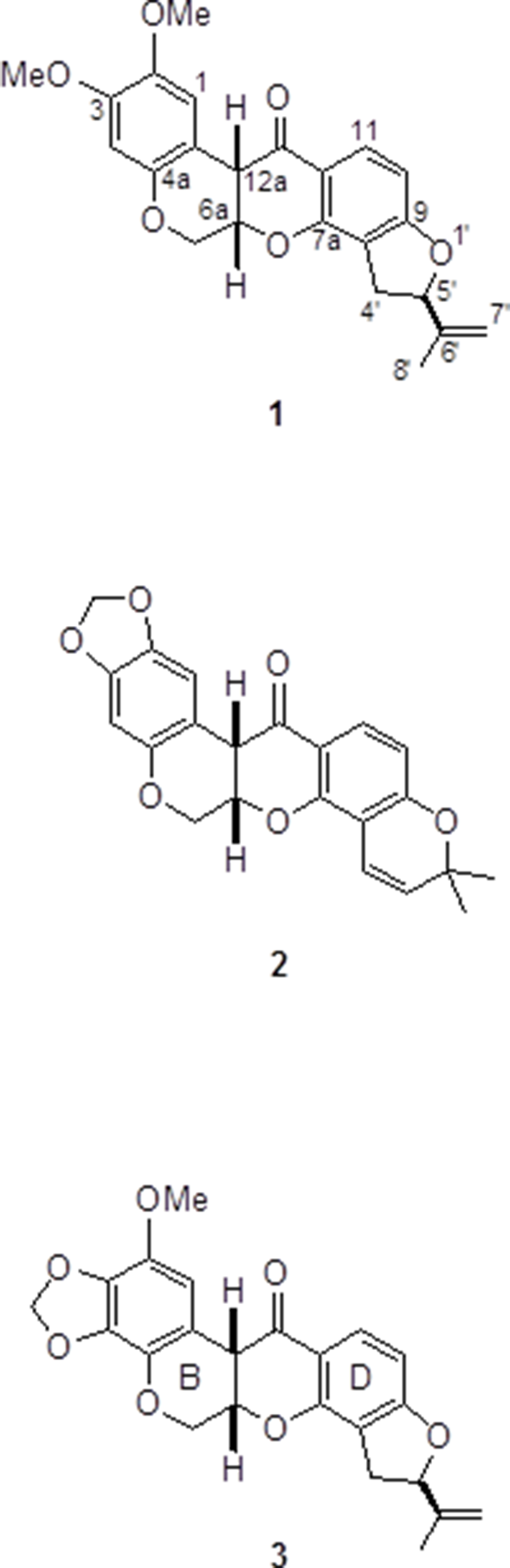
The bark of the roots of Piscidia carthagenensis afforded the known insecticides rotenone (1) and millettone (2), as well as the new rotenoid piscicartone (3). The structure of 3 followed from nuclear magnetic resonance studies, while its absolute configuration (AC) was determined by vibrational circular dichroism (VCD) measurements in comparison with discrete Fourier transform B3LYP/DGDZVPcalculated spectra using the CompareVOA software. In addition, the AC of 1 and 2 was verified using the same VCD methodology.
Piscidia carthagenensis Sarg. (family Fabaceae), popularly known as “barbasco de agua dulce,” is a tree that can reach heights of up to 15 m. It is native to tropical America, where it grows at altitudes between 10 and 1000 m above sea level.1-4 A decoction of the leaves has been used to impregnate horsetails and horsehair to eliminate infesting insects. The only report on biological activity of P. carthagenensis indicates that an ethanol extract of the branches and leaves showed very good activity in the brine shrimp lethality test.5
A study of the leaves of the tree, performed a few years ago,6 afforded the new compound 7,2′,5′-trimethoxy-3′,4′-methylene-dioxyisoflavone, as well as the known 6,7-dimethoxy-3′,4′-methylenedioxyisoflavone and 5,4′-dihydroxy-7,2′,5′-trimethoxy-isoflavone. We have now studied the bark of small sections of the roots of the same individual tree from which we have isolated the two known insecticides rotenone (1) and millettone (2), together with a new rotenoid, for which we propose the name piscicartone. Its structure elucidation and absolute configuration determination are described herein.
Extensive chromatographic separation of the chloroform-5% methanol fraction of the finely ground bark led to the isolation of rotenone (1), the identity of which was suggested by comparison of its 1H and 13C NMR data with published values,7 which for the 1H spectra were within 0.08 ppm and for the 13C spectra within 0.3 ppm, except for the C-12 carbonyl signal, which differed by 1.2 ppm, probably due to different concentrations used for the literature and current measurements; carbonyl groups are prone to associate with the solvent. Verification of the identity of 1 was made by direct comparison with an authentic commercial sample of rotenone, which showed no depression in melting point on admixture with the compound isolated; the IR spectra and optical rotations were also the same. In a similar way, compound 2 was identified as millettone (2) by comparison of its melting point, specific rotation, and spectroscopic data with literature values.8,9
The structure of piscicartone (3) could be evidenced using the 1H and 13C NMR data summarized in Table 1, and by comparison with those for rotenone (1) using very similar concentrations and instrumental conditions. The 13C NMR chemical shifts of C-6, of all carbon atoms of the C, D, and E rings, as well as those of the isopropylidene group of 3 are essentially identical to those of 1. A similar comparison of the 1H NMR data holds for the C, D, and E rings, and the isopropylidene group, although the two hydrogen atoms at C-6 are slightly shifted to higher frequencies. The main difference between 1 and 3 is that, in the former, the A ring holds two methoxy groups and two para distributed hydrogen atoms, while 3 shows signals due to one methylenedioxy group and one methoxy group, and therefore shows only one hydrogen atom signal. Thus, only six structural possibilities for 3 remain, which are the three possible locations for the methylenedioxy group, each one allowing two possibilities for the methoxy group and hydrogen atom placement. Careful inspection of the 1H NMR spectrum of 3, determined under very good magnetic homogeneity conditions, revealed the sole aromatic hydrogen atom signal as a long-range coupled doublet (J = 1.2 Hz), which, in principle, regardless of knowing the precise interaction, can only be understood when placing it at C-1, since otherwise it would be too far from any other hydrogen atom to provide a doublet of this magnitude. This reasoning leaves only two alternative structures for 3, by locating the methoxy group either at C-2 or C-4. The former possibility was confirmed from a gHMBC experiment, which revealed long-range correlations of H-1 at δ 6.44 with the carbon signals at δ 44.7 (C-12a), 108.9 (C-12b), 132.8 (C-4a), 136.4 (C-3), and 138.6 (C-2), in combination with the correlation of the hydrogen signal of the methoxy group at δ 3.77 with C-2 at δ 138.6.
Nuclear Magnetic Resonance Data Comparison of Rotenone (1) and Piscicartone (3).
bCoupling constants are given in the Experimental section.
A complementary structure confirmation for 3 was gained after PERCH10 simulation of the complete 1H NMR spin-spin system, as was recently reported for monoterpenes derived from ocimene11 and for cholesterol.12 The data revealed that the 1.2 Hz coupling constant is due to a 4J1,12a interaction, which is not directly evident by inspection of the spectrum since H-12a appears as a broadened doublet 3J6a,12a = 3.8 Hz, as H-12a also produces a long-range couplet with H-6A. Although PERCH simulations provide five digits after the decimal point for chemical shifts and four digits for coupling constants, in the present case the chemical shifts given in Table 1 have been rounded up to two digits and those for the coupling constants to one digit. The overall spectrum simulation converged to a very good 0.041%.
Regarding the absolute configuration (AC) of 3, it can be assumed that since the three isolated rotenoids share a common biogenetic origin, they should have the same AC, and that a relatively easy way of testing this would be by comparison of the experimental and calculated vibrational circular dichroism (VCD) spectra, since inspection of the molecular structures of 1–3 suggest they would have quite rigid molecular scaffolds and, therefore, relatively few low-energy conformers. This methodology has been used successfully13,14 for the AC determination of several types of natural products, although not yet for rotenoids.
The VCD studies were started after software construction of the molecular models to perform molecular mechanics conformational searches using the Monte Carlo protocol, as implemented in the Spartan 04 package. In the case of rotenone (1) the initial molecular mechanics force field (MMFF94) search rendered 35 conformers in a narrow energy gap of 1.16 kcal/mol, which after single-point discrete Fourier transform (DFT) optimization using the same software suit left only three conformers in an 1.08 energy gap, accounting for 95.0% of the total conformational distribution, the next conformer being 2.45 kcal/mol above the global minimum. In severe contrast, in the case of millettone (2), the MMFF calculation provided only two conformers in a very narrow range of 0.18 kcal/mol, which after single-point calculations expanded to a 1.26 kcal/mol energy gap. The same procedure, when applied to piscicartone (3), initially provided 14 conformers in a 10.83 kcal/mol energy gap, which were submitted to single-point energy optimization using DFT at the B3LYP/6-31G(d) level of theory with the same software. The outcomes of these calculations were six conformers, but now in a 2.51 kcal/mol energy gap accounting for 94.2% of the total conformational distribution, the next conformer being 3.13 kcal/mol above the global minimum. The three conformers of 1, the two conformers of 2, and the six conformers of 3 were further optimized at the B3LYP/DGDZVP level of theory using the Gaussian 09 suit to provide the Boltzmann distributions and relative energies given in Table 2. The final conformational optimization, IR and VCD calculations, at the same level of theory, were also performed using the Gaussian 09 software, and the pertinent thermochemical parameters, also summarized in Table 2, show the 11 final conformers for rotenoids 1 to 3.
aRelative to 1c (108.94 kcal/mol), 2a (94.30 kcal/mol), and 3c (478.29 kcal/mol).
bCalculated according to ΔE≅ −RT ln K.
cRelative to 1a (−841 478.47 kcal/mol), 2a (−816 070.61kcal/mol), and 3a (−887911.23 kcal/mol).
dRelative to 1a (−841 306.47 kcal/mol), 2a (−815 923.80kcal/mol), and 3a (− 887 763.39 kcal/mol).
eRelative to 1a (−841 357.94kcal/mol), 2a (−815 969.81kcal/mol), and 3a (−887 814.24kcal/mol).
fCalculated according to ΔG = −RT ln K.
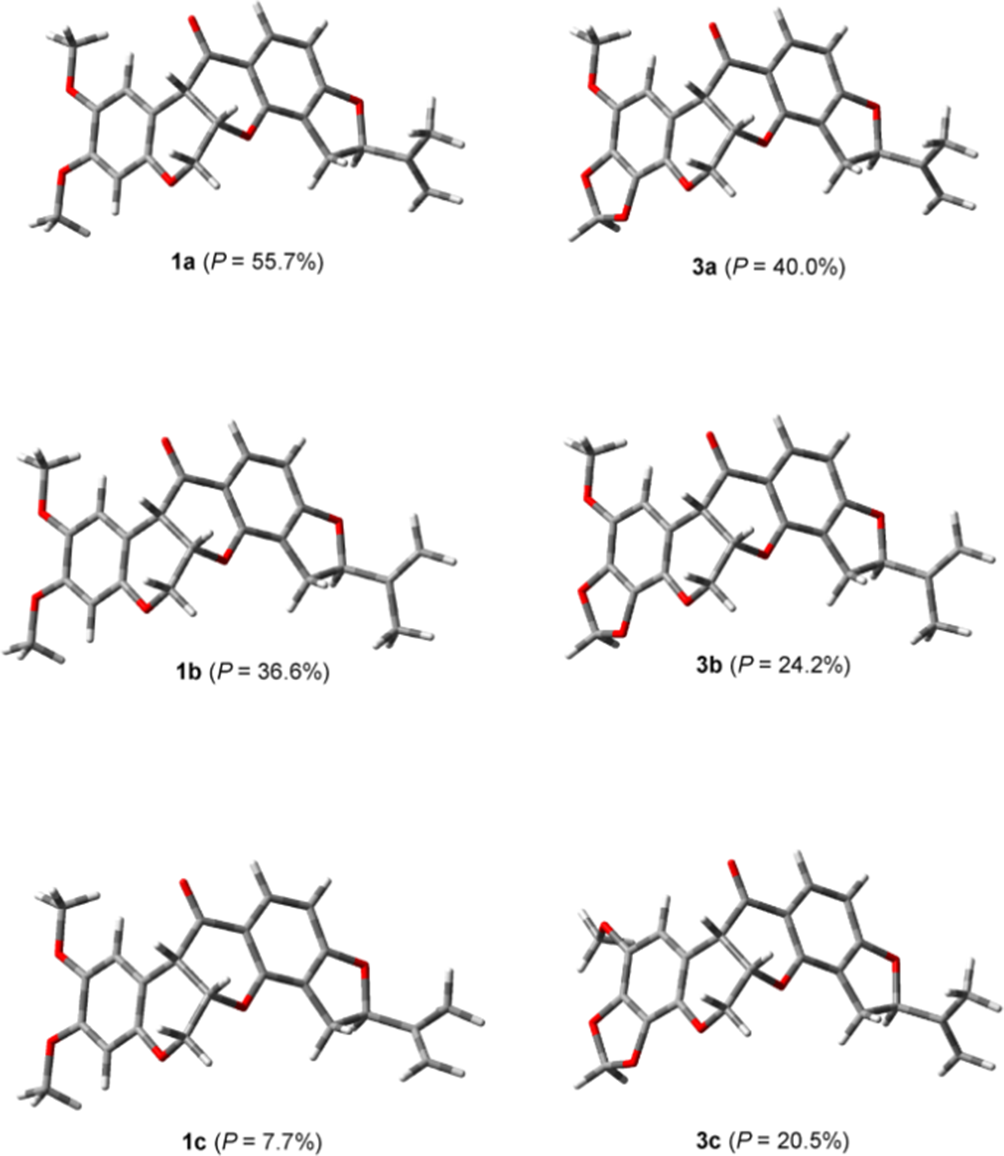
Comparison of the calculated and experimental spectra using the Compare VOA software15 provided the statistical data shown in Table 3. The overall comparisons were very good due to the molecular rigidity of the rotenoids, which, therefore, provided quite sharp IR and VCD absorption bands, as can be observed in Figures 1–3 for 1to 3, respectively. In turn, inspection of the 3 low-energy conformers for 1 and 3, shown in Figure 4, reveals that the scaffold of these rotenoids is quite rigid. In the case of 1 the conformers differ only in the isopropylidene group orientation, as evidenced by the C-4′–C-5′–C-6′–C-7′ dihedral angles for conformers 1a to 1c, which are 111.2°, −111.9°, and 17.8°, respectively. In the case of 3 there is also a change in the methoxy group orientation. Thus, the C-4′–C-5′–C-6′–C-7′ dihedral angles for conformers 3ato 3c are 111.4°, −112.0°, and 110.7°, respectively, while the C-1–C-2–O–Me dihedral angles for these conformers are 0.1°, 0.0°, and 132.4°, respectively.
Comparison of the experimental IR and vibrational circular dichroism spectra of (–)-rotenone (1) with the discrete Fourier transform spectra calculated at the B3LYP/DGDZVP level of theory.
Comparison of the experimental IR and vibrational circular dichroism spectra of (–)-millettone (2) with the discrete Fourier transform spectra calculated at the B3LYP/DGDZVP level of theory.
Comparison of the experimental IR and vibrational circular dichroism spectra of (–)-piscicartone (3) with the discrete Fourier transform spectra calculated at the B3LYP/DGDZVP level of theory.
The 3 low-energy conformers of (left) rotenone (1) and of (right) piscicartone (3).
Confidence Level Data for the Infrared and Vibrational Circular Dichroism Spectra Comparison of 1 to 3.
cVCD spectral similarity for the correct enantiomer in percentage.
dVCD spectral similarity for the opposite enantiomer in percentage.
eEnantiomer similarity index, calculated as the SE − S−E difference.
fConfidence level for the absolute configuration determination in percentage.
The discovery of piscicartone (3) could lead to the development of new insecticidal molecules since 3 possesses one of the methoxy groups of rotenone (1) and it is known16 that 3,4-methylenedioxyphenyl compounds are excellent synergists for pyrethroids, and enhanced insecticidal activity has been observed when these 3,4-methylenedioxyphenyl compounds are applied in combination with 1.17 Thus, the combined effect of rotenoids 1 to 3 in the bark of the roots of the studied tree seems to provide a protection barrier for this tree growing in the hot and humid landscapes of the coastal regions of the State of Oaxaca, Mexico.
Experimental
General
Melting points were determined on a Fisher-Johns apparatus and are uncorrected. Optical rotations were determined from EtOH solution at 25°C using a Perkin-Elmer 341 polarimeter and IR and VCD measurements were made on a BioTools dualPEM ChiralIR FT spectrophotometer. NMR measurements were obtained from CDCl3 solutions containing TMS at 300 MHz for 1H and 75 MHz for 13C on a Varian Mercury spectrometer; the HRMS was measured on a Waters Synapt G2 spectrometer at the Department of Chemistry and Biochemistry, University of Colorado, Boulder, CO, USA. column chromatography was carried out on Merck silica gel 60 (230, 400-mesh ASTM) and TLC was developed on silica gel 60 F254 plates.
Plant Material
The studied exemplar of Piscidia cartagenensis, located in the neighborhood of Guelaguichi, some 17 km southwest (N 16° 07′ 02″, W 99° 17′ 50″) of the city of Salina Cruz, State of Oaxaca, Mexico, is the same individual tree from where we collected the leaves that provided 7,2′,5′-trimethoxy-3′,4′-methylenedioxyisoflavone.6 Small portions of the bark were removed from parts of the roots of the tree during April 2017.
Rotenoids Isolation
A total of 320 g of the dried and finely ground bark was placed in a glass column of 8 cm outside diameter to provide a cylinder of material measuring 10 cm in height, which was extracted with chloroform containing 5% methanol to provide 12 fractions each of 100 mL. Upon evaporation, fractions 6 to 9 provided semicrystalline residues. These were combined to a total 70 mg of residue, which was carefully column chromatographed on silica gel and eluted with hexanes with increasing proportions of CH2Cl2. The earlier fractions, eluted with hexanes-CH2Cl2 3:1, provided 5.6 mg of millettone (2), followed by 25.4 mg of the same compound admixed with some unidentified contaminants. The fractions eluted with hexanes-CH2Cl2 2:1 provided 5.6 mg of piscicartone (3), and finally the fractions eluted with hexanes-CH2Cl2 1:1 gave 10.6 mg of rotenone (1).
Samples of 3.8 mg of 1, 4.3 mg of 2, and 4.0 mg of 3 were individually dissolved in 150 µL of 100% atom-D CDCl3 and placed in spectrophotometer cells with BaF2 windows and a path length of 100 µm. The VCD data were acquired at a resolution of 4 cm-1 for 6 hours, and the stability of each sample was verified by 1H NMR measurements immediately before and after the VCD determination. A baseline correction was performed by subtracting the spectrum of the solvent acquired under identical instrument conditions.
Vibrational Circular Dichroism Calculations
The initial conformational searches for 1 to 3 were started generating each molecular model in Spartan 04 software (Wavefunction, Inc., Irvine, CA, USA). Preliminary optimization using the Monte Carlo protocol and the MMFF94 force field afforded 35, 2, and 14 conformers in 1.16, 0.18, and 10.83 kcal/mol energy windows for 1 to 3, respectively. Each set of conformers was then submitted to single-point energy calculations using DFT and the B3LYP/6–31G(d) functional and basis set. This left 3, 2, and 6 conformers in 1.08, 1.26, and 1.51 kcal/mol energy gaps for 1 to 3, respectively. All conformers were completely optimized using Gaussian 09 (Gaussian, Inc., Wallingford, CT, USA) at the DFT and B3LYP/DGDZVP level of theory and their frequencies were calculated at the same level of theory. After complete optimization, for the calculation of final vibrational normal modes and rotational strengths, the same functional and basis set were used, thus generating the final calculated IR and VCD spectra considering Boltzmann distributions based on their ΔG values. The thermochemical analysis was made at 298 K and 1 atm, and the VCD and IR frequencies were plotted using Lorentzian band shapes and bandwidths of 6 cm-1. Calculated vibrational frequencies were obtained from the most stable conformer using the GaussView 5.0 software. DFT calculations required on average 12 hours computational time per conformer when using a PC operating at 3 GHz with 4 Gb RAM. In turn, calculated and experimental spectra were compared using the CompareVOA (BioTools, Jupiter, FL) software.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partial supported by CONACYT-Mexico grant number 284194.
References
1.
SousaM.MedinaR.AndradeG.RicoLM. Leguminosas. In: García-MendozaAJ.OrdóñezMJ.Briones-SalasM., eds.Biodiversidad de Oaxaca. Mexico: Instituto de Biología UNAM-Fondo Oaxaqueño para la Conservación de la Naturaleza-World Wildlife Found; 2004:249-269.
GuerreroRO.RiveraSM.RiveraS.SueiroLA. Bioassay screening of Amazonian plants. Puerto Rico Health Sci J. 2003;22:291-297.
6.
OrtegaAR.ToscanoRA.Hernández-BarragánA.Alvarez-CisnerosC.Joseph-NathanP. Structure elucidation of a new isoflavone by exclusive use of 1H NMR measurements. Magn Reson Chem. 2015;53(10):860-865.doi:10.1002/mrc.4278
7.
BlaskóG.ShiehHL.PezzutoJM.CordellGA. 13C-NMR spectral assignment and evaluation of the cytotoxic potential of rotenone. J Nat Prod. 1989;52(6):1363-1366.doi:10.1021/np50066a035
8.
OllisWD.RhodesCA.SutherlandIO. The extractives of Millettia dura (Dunn). The constitutions of durlettone, durmillone, milldurone, millettone and millettosin. Tetrahedron. 1967;23:4741-4760.
9.
YenesewA.MidiwoJO.WatermanPG. 6-Methoxycalpogonium Isoflavone A: A New Isoflavone from the Seed Pods of Millettia dura. J Nat Prod. 1997;60(8):806-807.doi:10.1021/np9605955
10.
LaatikainenR.TiainenM.KorhonenS-P.NiemitzM. (Eds).
Computerized analysis of high-resolution solution-state spectra. In: Encyclopedia of Magnetic Resonance. Harris RK, Wasylishen RE. Chichester: John Wiley; 2012:677-688.
11.
JulioLF.Burgueño-TapiaE.DíazCE.Pérez-HernándezN.González-ColomaA.Joseph-NathanP. Absolute configuration of the ocimene monoterpenoids from Artemisia absinthium. Chirality. 2017;29(11):716-725.doi:10.1002/chir.22741
12.
Pérez-HernándezN.Becerra-MartínezE.Joseph-NathanP. Complete 1H NMR assignment of cholesteryl benzoate. Steroids. 2018;138:72-81.doi:10.1016/j.steroids.2018.06.010
13.
Joseph-NathanP.Gordillo-RománB. Vibrational circular dichroism absolute configuration determination of natural products. In: KinghornAD.FalkH.KobayashiJ., eds.Progress in the Chemistry of Organic Natural Products. Cham, Switzerland: Springer International Publishing; 2015:311-451.
14.
Burgueño-TapiaE.Joseph-NathanP. Vibrational circular dichroism: recent advances for the assignment of the absolute configuration of natural products. Nat Prod Commun. 2015;10(10):1785-1795.doi:10.1177/1934578X1501001036
15.
DebieE.De GussemE.DukorRK.HerreboutW.NafieLA.BultinckP. A confidence level algorithm for the determination of absolute configuration using vibrational circular dichroism or Raman optical activity. ChemPhysChem. 2011;12(8):1542-1549.doi:10.1002/cphc.201100050
16.
BerozaM. Identification of 3,4-Methylenedioxphenyl synergists by reversed-phase paper chromatography. Anal Chem. 1956;28(10):1550-1552.doi:10.1021/ac60118a015
17.
MitanoM. Rotenoids. XXII. Total synthesis of isomillettone. J Org Chem. 1970;35:245-249.