Clerodane diterpenoids isolated from the plant kingdom account for the majority of diterpenoids. They include more than 1300 compounds possessing a wide variety of functional groups and stereochemistry on decaline cores. Some compounds possess fascinating biological as well as pharmacological activities such as insect antifeedent, opioid receptor agonist, nerve growth factor (NGF)-potentiating, anti-ulcer, cytotoxic, anti-inflammatory, antiparasitic, antifungal, antibacterial, antitumor, and antibiotic activities. These aspects were reviewed comprehensively by Lee et al covering the papers from 1990 to 2015,1 in which the distribution, chemotaxonomic significance, chemical structures, and biological activities of clerodane diterpenoids (clerodanes) were summarized, along with structure–activity relationship (SAR) correlations and mode of action of active clerodanes.
Basic framework of clerodanes having neo clerodane absolute configuration.
Relative stereochemistry of clerodanes is classified into four categories according to the 5:10 and 8:9 positions, in which the trans-5:10 clerodanes 1 and 2 predominate in nature (Figure 1).1
Opposite absolute stereochemistry had been assigned until the stereochemistry of clerodin (6) was revised in 1979.2–5 In order to avoid confusion, the absolute stereochemistry of clerodanes determined after the revision was indicated by the prefix neo- or ent-neo- for enantiomers (Figure 2).
Absolute configuration of clerodanes.
Carbons 12 to 16 are oxidized usually to diene, furan, lactone, or hydrofurofuran, which gives characteristic structural features to clerodanes.
The biosynthetic pathway to trans-clerodane has been proposed in which enzymatic cyclization of geranylgeranylpyrophoshate (7) (GG-OPP) leads to labdane-type cation eight and the subsequent concerted migration of hydrides and methyl groups provides trans-clerodanes. The formation of cis-clerodane, which occupies almost one-fourth of the clerodane family, suggests a minor stepwise pathway via cation 9 (Scheme 1).1,6
Biogenetic pathway toward clerodanes.
The diversity of structures and biological activities has enhanced total syntheses of clerodanes, which were compiled so far in the review by Tokoroyama in 20007 and Ohsaki in 2004.6 The present review aims to fill the gaps in the compilation up to 2017.
For the construction of decaline cores, a wide variety of processes have been employed, such as Diels–Alder reaction, ene reaction, alkylation, Dieckman condensation, and Michael reaction, either in an inter- or intramolecular manner. Syntheses of the cis-decaline core were accomplished by intermolecular Diels–Alder reaction. Wieland–Miescher ketone analog 157 (Scheme 17) and 176 (Scheme 19) are compatible for syntheses of trans-clerodane natural products owing to the easy availability of enantiomerically enriched compound,8 along with the established protocol to introduce three successive stereogenic centers at C5, C9, and C10 by reductive alkylation. How to construct decaline cores and to introduce oxygenated functionalities stereoselectively are key to the success of total syntheses. A biogenetic-type approach is not known probably due to the complex structures of clerodanes.
Several clerodanes bearing spiro-γ-lactone moieties at C-9 were known, such as teucvin (28) and montanin (36) (Scheme 3). Toward total syntheses of these compounds, Ley et al constructed spiro-decaline framework 13 employing the intermolecular Diels-Alder reaction between silyldienolether 11 and (Z)-α-ethylidene-γ-butyrolactone 12 as a key step, which was carried out without a solvent in a presilylated glass pressure vessel to give adduct 13 (Scheme 2).11 Starting materials were recovered when the reaction was carried out in the presence of a Lewis acid. After phenylselenylation of the adduct 13, enones 15 and 16 were obtained.
Syntheses of spiro-lactone clerodane cores 15 and 16.11
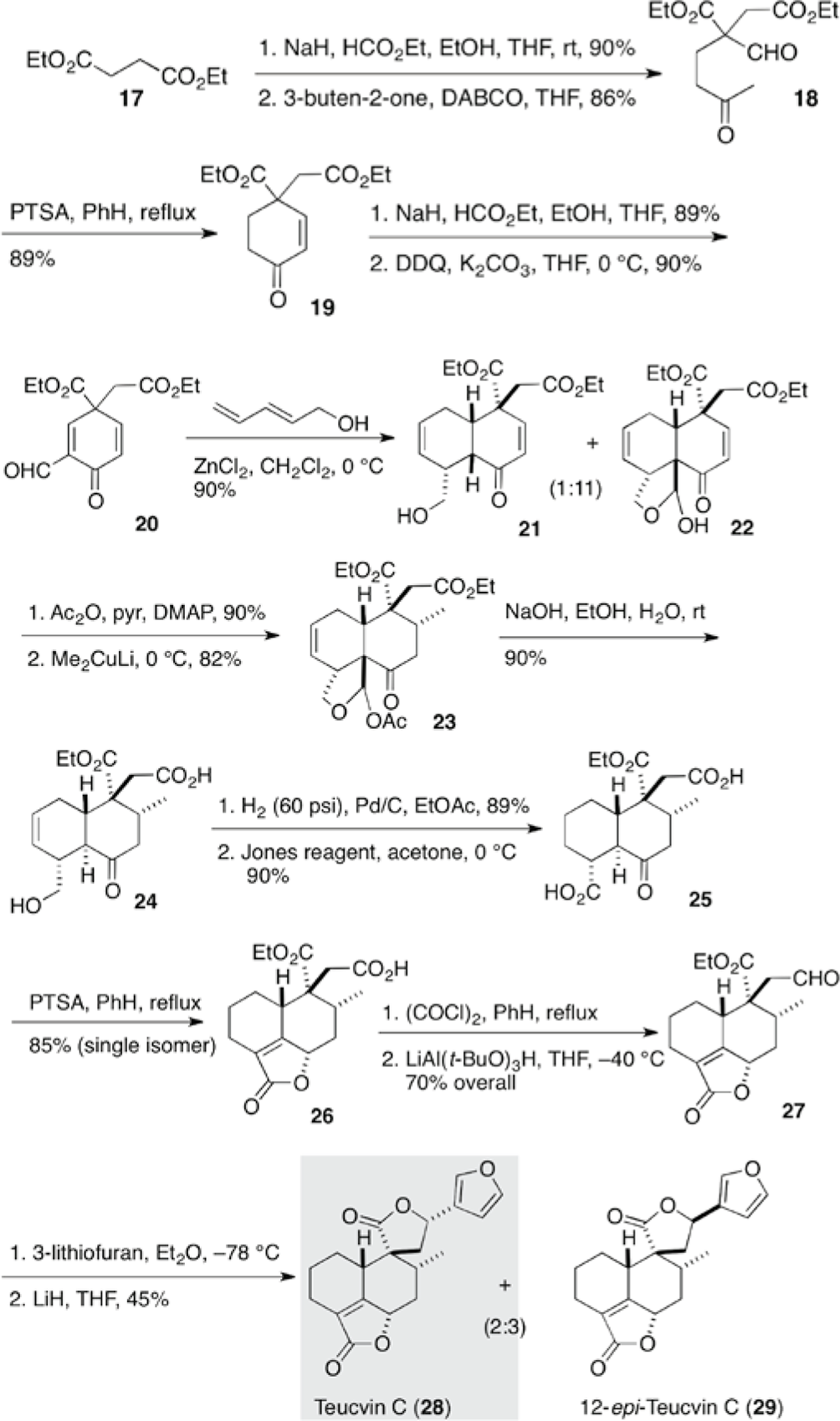
Teucvin C (28) and 12-epi-teucvin (29) (Scheme 3), 19-nor-γ-lactone clerodanes, isolated from Teucrium viscidum Blume,12 have four stereogenic centers on a decaline core attached to an α,β-unsaturated-γ-lactone unit and a spiro γ-lactone moiety containing a pendant furyl group. Teucvin C (28) showed amebicidal and root growth inhibitor activity. Liu et al reported the first total synthesis of racemic teucvin C (28) and 12-epi-teucvin C (29), based on the spiro-decaline synthesis by intramolecular Diels–Alder reaction (Scheme 3) as a part of their efforts to synthesize spiro-γ-lactone clerodanes.11 The Diels–Alder reaction of cyclohexadienone 20 and trans-2,4-pentadien-1-ol was followed by hemi-acetalization to give decalone 22 as a major component. The dienophile 20 was derived from diethyl succinate (17) in five steps. The addition of the pentadien-1-ol proceeded selectively from the same side of the quaternary ester group. Conjugate addition of a methyl group proceeded selectively from the less hindered α-face of the molecule to give decalone 23. Alkaline treatment of 23 resulted in the hydrolysis of the acetyl group, removal of the resulting formyl group, and epimerization at the ring juncture to trans, which was followed by regioselective hydrolysis of the exposed ester to provide hydroxy-acid 24. Addition of 3-furyllithium to aldehyde 27 furnished racemic teucvin C (28) and 12-epi-teucvin C (29).
Total syntheses of (±)-teucvin C (28) and 12-epi-teucvin C (29).12
Montanin A (36) was isolated from the aerial part of Teucrium montanum L. as a part of a biogenetic congener of the Teucrium clerodane family (Scheme 4). Teuscorolide (40) was extracted from the aerial part of Teucrium scorodonia L. (Labiatae), which has an extra double bond at C6-C7 of teucvin C (28). The plant is used as a folk medicine to treat skin afflictions, diseases of the blood, fever, and cold.
Total syntheses of (±)-montanin A (36) and (±)-teuscorolide (40).13,14
Liu et al reported the total synthesis of racemic montanin A (36) and teuscorolide (40) (Scheme 4).13,14 Controlled treatment of teucvin C synthetic intermediate 23 in the previous paper11 with base resulted in selective hydrolysis of the acetate, elimination of the formyl group, and isomerization of the ring juncture successively. After transformation to keto-carboxylic acid 31, butenolide 32 was obtained by acid treatment via the formation of an enol lactone and the double bond isomerization. Reduction with more than three equivalents of DIBAL-H transformed the butenolide to the furan and the exposed ester selectively to primary alcohol to provide hydroxyfuran 33. The oxidation of 33 was accomplished only by Ag2CO3/celite to give an aldehyde. Addition of 3-lithiofuran and subsequent lactonization provided montanin A (36). PDC oxidation of montanin A (36) gave γ-hydroxy butenolide 39. The reaction is supposed to proceed via [4 + 2]cycloaddition of furan with Cr(II)=O, formation of γ-keto-α,β-unsaturated aldehyde, oxidation, and ring closure, in which only the strained and electron rich furan at C4-C6 reacted leaving the furan at C12 intact. Dehydration with acid completed the synthesis of teuscorolide (40).
At the same time, air oxidation of montanin A (36) led to teucvin C (28) (Scheme 5). Radical abstraction from CHCl3 at C6 is assumed to proceed at the β-face giving acetal 42 stereoselectively.
Oxidation of montanin A (36) to (±)-teucvin C (28).13,14
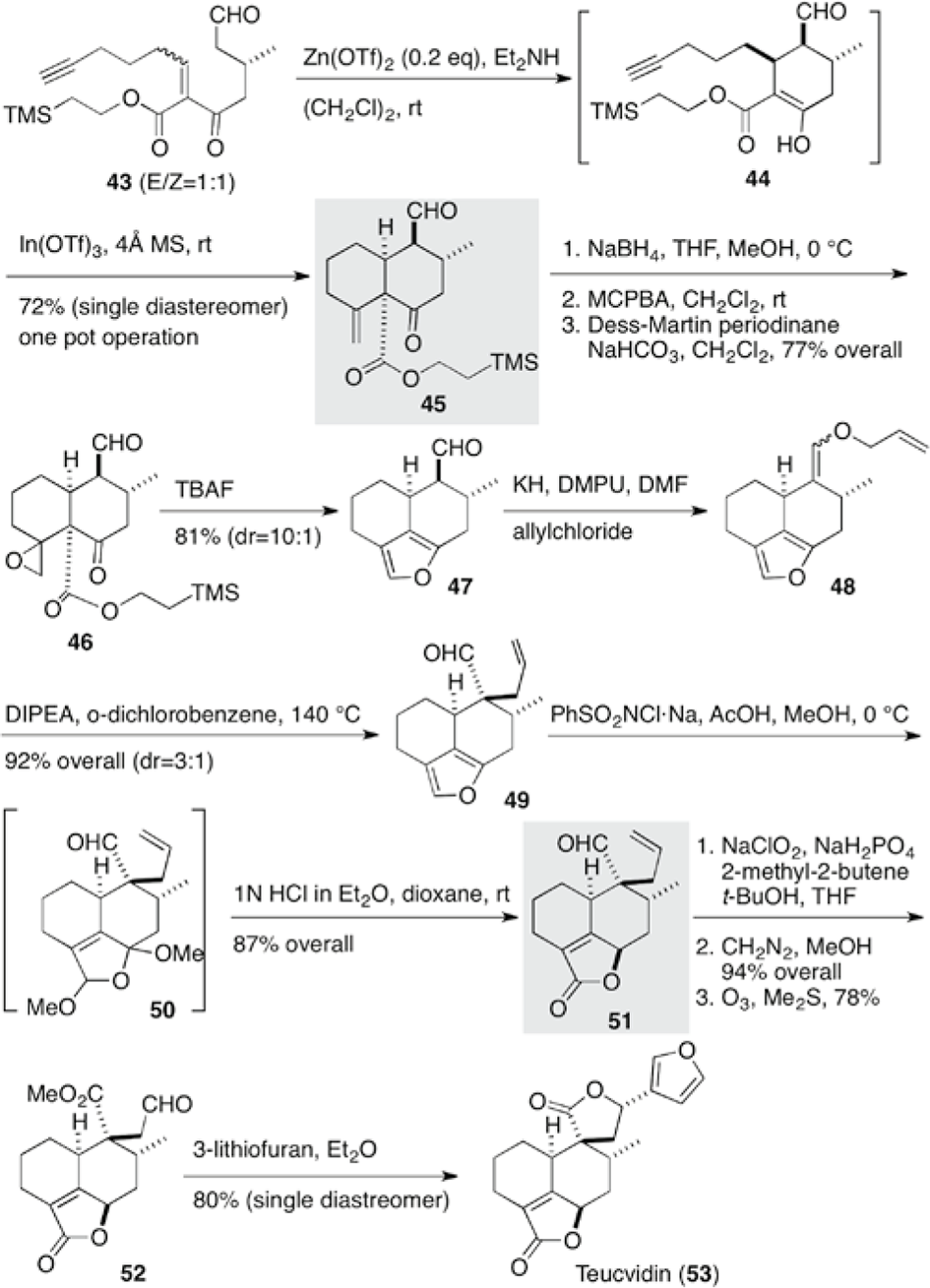
Teucvidin (53), an epimer at C10 of teucvin C (28), was isolated from Teucrium viscidum Blume var. Miquelianum. Lee et al accomplished the total synthesis of (−)-teucvidin (53) based on sequential Michael/Conia ene reactions being key to construct the cis-decalone framework 45 (Scheme 6).15 Treatment of (E)/(Z) mixture (1:1) of (−)-aldehyde 43 with Zn(OTf)2 promoted the stereoselective intramolecular Michael addition via twisted chair-like transition state of the (E)-aldehyde 43. Conia ene reaction of the resulting β-keto-ester 44 proceeded with the addition of In(OTf)3 in one pot to provide, as a single diastereomer, cis-decalone 45, which bore the requisite three stereogenic centers and functional groups. (Z)-Aldehyde 43 is easily isomerized to (E)-aldehyde 43. In order to avoid Baeyer-Villiger reaction, the ketone at C6 was reduced prior to epoxidation and the hydroxyl group was oxidized back. Without isolation, treatment with TBAF resulted in dealkoxycarbonylation to give an enolate, which triggered opening of the epoxide at C-4, followed by subsequent cyclization and dehydration to introduce the furan moiety at C4-C6. Although direct allylation to the α-position of aldehyde 47 was not successful, Claisen rearrangement of allyl ether 48 furnished α-allyl aldehyde 49 in a diastereomeric ratio 3:1 in favor of 49. The furan moiety was oxidized stereoselectively to butenolide 51 by PhSO2NCl·Na. Transformation of the formyl group at C-9 into an ester group and subsequent cleavage of the allyl group, followed by stereoselective addition of 3-lithiofuran, completed the total synthesis of (−)-teucvidin C (53) in 22% overall yield in 12 steps. The higher stereoselectivity of the furan addition might be due to chelation of 3-lithiofuran between the formyl and the ester groups.
Teucrolivin (67) was isolated from the aerial parts of Teucrium oliverianum, a plant native to Saudi Arabia, which finds use as a folk medicine for the treatment of diabetes. Ams and Barriault reported the synthetic study of teucrolivin (67) employing sequential oxy-Cope/Claisen/ene reactions as a key step (Scheme 7).16 An isopropenyl group was introduced via 1,2-diaxial ring opening of epoxide 56, which was followed by protection of the diol and subsequent oxidation to give ketone 58. Stereoselective addition of (E)−2-lithio-2-butene to ketone 58 led to alcohol 59. O-allylation with allylbromide 60 followed by microwave heating at 220°C promoted the oxy-Cope reaction leading to a 10-membered ring 62. Subsequent [3,3]Claisen reaction, followed by transannular ene reaction of 63, furnished decaline 64, which has four contiguous desired stereogenic centers. Cleavage of the double bond followed by β-elimination of the carbonate ester provided the synthetic precursor 66 of teucrolivin (67) in 16 steps.
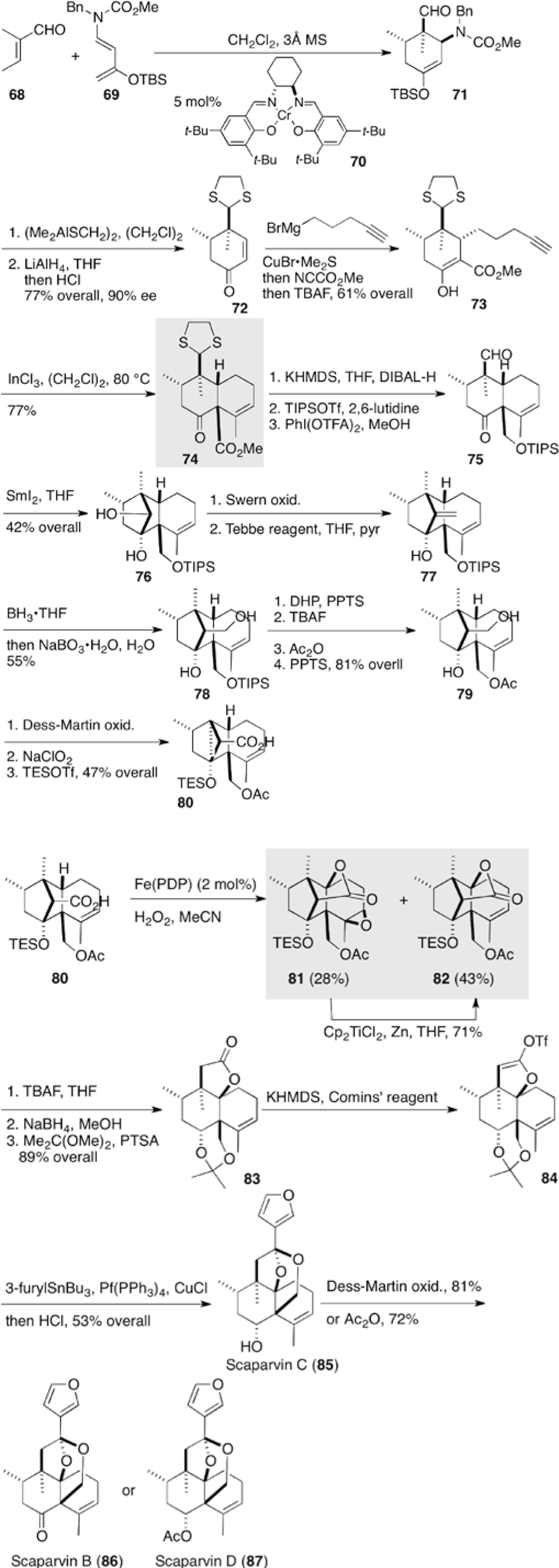
Scaparvins (85-87), isolated from the epilithic liverwort Scapania parva Steph., possess a distinct cage, including an intramolecular ketal, along with 5 to 6 contiguous stereocenters (Scheme 8). These compounds have similar functionalities as teucrolivin A (67), though the ring junctures are trans and antipodal. Total syntheses of these compounds 85-87 have been accomplished by Snyder et al via construction of cis-decaline core 74 by Conia ene reaction, followed by selective C–H functionalization, leading to γ-lactone 82 and subsequent acetalization (Schemes 8; 9).17
Total syntheses of scaparvins B (86), C (85), and D (87).17
The enantioselective Diels–Alder reaction of a Rawal diene 69 with tiglic aldehyde (68) in the presence of a chromium chiral salen complex 70 provided enol–ether 71 in 90% ee. Dithiolane protection, followed by reduction of the carbamate, led to cyclohexenone 72. In the subsequent conjugate, addition of a Grignard reagent proceeded on the α-face of 72 as a result of shielding by coordination of copper on the β-face. Trapping the enolate with Mander’s reagent gave keto-ester 73. The Conia ene reaction mediated by indium chloride proceeded to give cis-decalone 74 in a similar manner as shown in the synthesis of teucvidin (53) (Scheme 6),15 accompanying isomerization of the exo-methylene group at C4. After reduction of the ester at C5 and deprotection/protection sequence, SmI2-mediated pinacol coupling of the resulting keto-aldehyde 75, which transformed into bridged diol 76. Swern oxidation followed by Tebbe olefination led to bridged exo-methylene 77, the hydroboration/oxidation of which proceeded from the outer face with complete stereocontrol. The bulky TIPS group was replaced by Ac to give alcohol 79 for the forthcoming C-H oxidation. Oxidation of the primary alcohol 79 provided a precursor 80 for the key C-H oxidation.
The key C-H oxidation of 80 was carried out according to the procedure by White et al19,20 to afford bridged lactone 82 and its epoxide 81, which was reduced back to the lactone 82 (Scheme 8). Deprotection of the TES group resulted in concomitant opening of the bridge, which was followed by the reduction of the ketone and protection to give lactone 83. Owing to difficulty with the introduction of the furyl unit directly leading to furyl ketone, the lactone 83 was transformed to vinyl triflate 84, whose palladium-catalyzed coupling with furyltin and subsequent hydrolysis furnished scaparvin C 85. Oxidation or acetylation finally furnished either scaparvin B (86) or D (87).
6β-Acetoxy-2-oxokolavenool (97) is a less common cis-clerodane and a constituent of several Mexican Stevia species. Liu et al synthesized 6β-acetoxy-2-oxokolavenool (97) via construction of decalone 92 by the face selective Diels–Alder reaction, which has requisite successive stereogenic centers for cis-clerodane compounds (Scheme 9).18 Synthesis of dienophile 91 started from 3-ethoxy-2-cyclohexenone 88. After α-formylation of the enone 89, formation of an isoxazole ring and its rearrangement introduced a cyano group to give α-cyanoketone 90, which was oxidized by DDQ to provide dienophile 91. The Diels-Alder reaction with trans-piperylene proceeded from the same side of the methyl group in the presence of ZnI2 to furnish cis-decalone 92. Treatment of the decalone 92 with lithium naphthalenide generated an enolate at C5, which was methylated to give cis-decalone 93 stereoselectively as a result of 1,3-steric interactions between the axial proton at C7 and two axial methyl groups at C4 and C9. 1,4-Conjugate addition of Me2CuLi with TMSBr proceeded stereoselectively from the α-face to give decalone 94. Keto-alcohol 96 was obtained via Wacker oxidation of the double bond of the side chain and subsequent addition of a vinyl group. Transformation of keto-alcohol 96 to 6β-acetoxy-2-oxokolavenool (97) is known.21,22
Total synthesis of (±)−6β-acetoxy-2-oxokolavenool (97).18
An alternative synthesis of 6β-acetoxy-oxokolavenool (97) from known enone 98 was reported via a similar route (Scheme 10).23
Total synthesis of (±)−6-acetoxy-2-oxokolavenool (97).23
7-Oxokolavenic acid (119) was isolated from an extract of the aerial parts of Platychaete aucheri. Kato et al utilized steric effects of the pinane framework as a chiral template for the synthesis of the trans-clerodane 119 (Scheme 11).24 Conjugate addition followed by methylation of (−)-verbenone (106) derived from (+)-nopinone gave ketone 107, which was transformed to bicyclic acetate 108. Opening of the 4-membered ring proceeded quantitatively to give enol-acetate 109, which was transformed to aldehyde 113 through several steps. Treatment with Et2AlCl promoted ene reaction to provide trans-decaline 114. Conjugate addition of the Grignard reagent to enone 115 proceeded stereoselectively to give adduct 116. Wacker oxidation of the terminal double bond and subsequent Horner–Wadsworth–Emmons reaction completed the first total synthesis of 7-oxokolavenic acid 119. Reduction and acetylation of methyl 7-oxokolavenate led to methyl solidagonate (120), isolated from the root of Solidago altissima.
Total syntheses of (−)−7-oxokolavenic acid (119) and methyl solidagonate (120).24
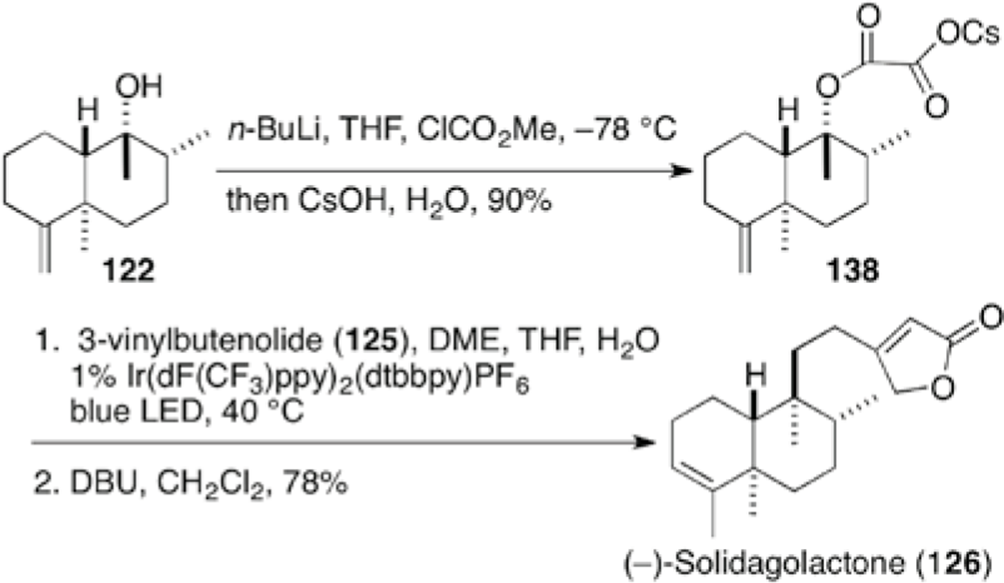
Solidagolactone (126), PL3 (127), and annonene (128) were isolated from the root of Solidago altissima, from the stem bark of Polyalthia cheliensis, and from bulbs of Annona coriacea, respectively. Overmann et al completed the total syntheses of these clerodanes, employing a 1,6-conjugate addition of organocopper reagent to 3-vinylbutenolide (125) (Scheme 12).25 Addition of MeMgBr to the known trans-decalone 12126 proceeded from the β-face to give alcohol 122, stereoselectively. After SN2 substitution of the hydroxyl group with sulfide 123, the exo-methylene at C-4 was isomerized to endo-olefin leading to 124. Tertiary organocopper reagent generated from the sulfide 124 with LiDBB and CuBr·Me2S added to 3-vinylbutenolide (125) in a 1,6-manner to give a mixture of solidagolactone (126) and its β,γ-double bond isomer, in which the addition proceeded stereoselectively on the equatorial side due to 1,3-steric repulsion between the methyl group at C5 and the axial proton at C-7. Treatment with DBU provided solidagolactone (126), which was in turn transformed into PL3 (127) by oxidation of its silylenol ether and into annonene (128) by reduction, respectively.
Total syntheses of (±)-solidagolactone (126), PL3 (127), and annonene (128).25
The same authors also accomplished the total synthesis of (−)-solidagolactone (126) by a 1,6-conjugate addition of a tertiary alkyl radical generated by visible light photoredox reaction as a key step (Scheme 13).25 After hydroalumination of chloroalkyne 129 with Ni catalyst, the resulting internal vinylaluminum intermediate was added to 3-methylyclohex-2-en-1-one (130) in a 1,4-manner in the presence of CuCl2 and Hoveyda chiral Ag-NHC ligand was added to furnish adduct 131 in 84% yield, enantioselectivity. Intramolecular alkylation led to decalone 132. Methylation at C-8, followed by introduction of a cyano group at C9 by TosMIC, provided nitrile 133. Methylation at C9 proceeded diastereoselectively from the β-face. Reduction of the nitrile 134 and subsequent Pinnick oxidation gave carboxylic acid 135. After transformation of the carboxy group to a N-hydroxyphthalimide group, the 1,6-conjugate coupling reaction with 3-vinylbutenolide (125) under irradiation of blue LED in the presence of Hantzsch ester as a reductant proceeded, also from the β-face of the molecule, to give an adduct and its β,γ-double bond isomer. Isomerization into butenolide 137 by DBU and subsequent isomerization of the exo-methylene group at C4 by Rh catalyst completed the total synthesis of (−)-solidagolactone (126).
Furthermore, the total synthesis of (−)-solidagolactone 126 was refined in six steps from known decalone 122 employing cesium oxalate 138 as a photoredox precursor without the addition of Hantzsch ester (Scheme 14),27 in which the iron (III) catalyst was optimal as a photoredox catalyst. The present protocol is promising towards the future syntheses of a number of clerodane diterpenoids.
More compact synthesis of (−)- solidagolactone (126).27
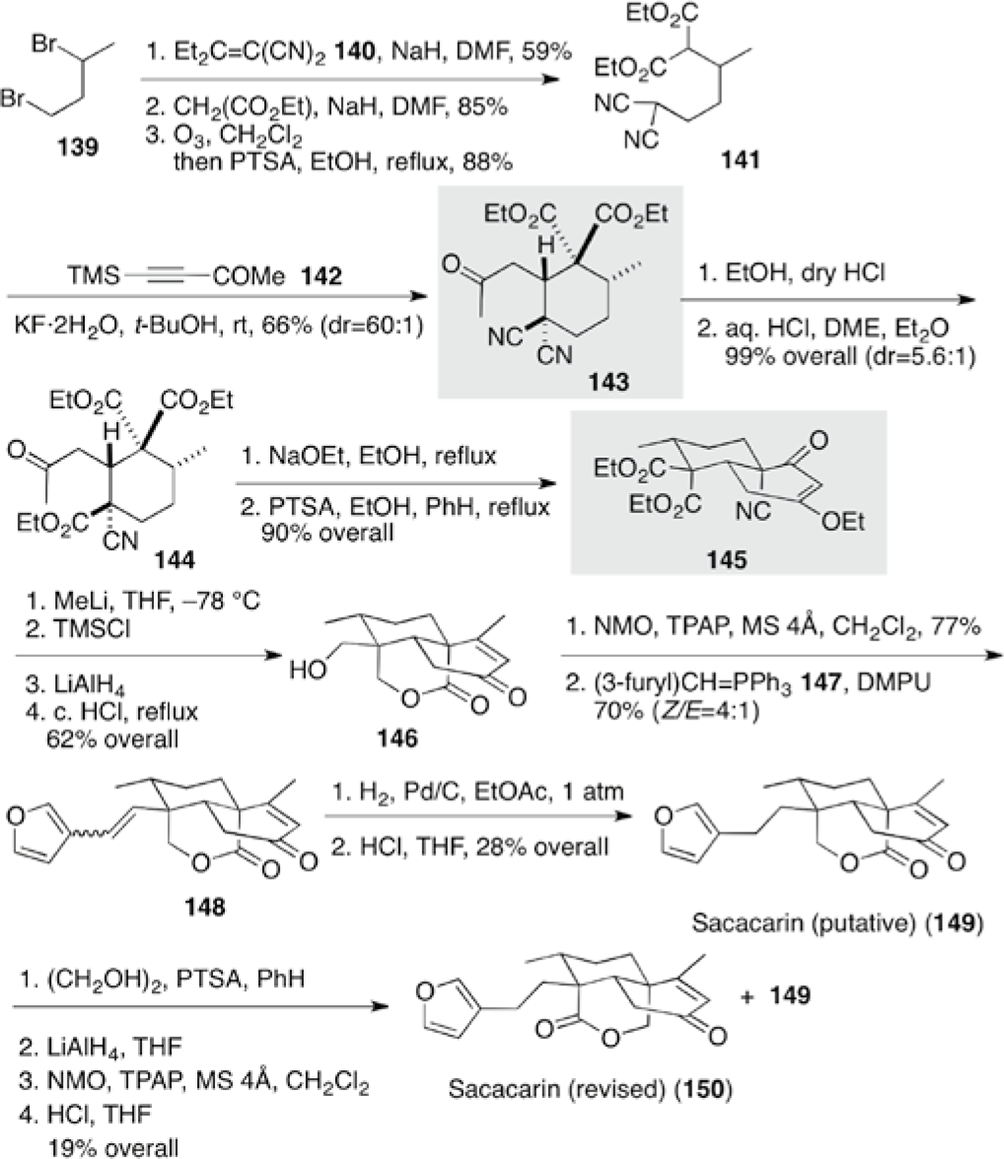
Sacacarin (150) was isolated as a minor constituent of the bark of a Brazilian tree, which was used as a folk medicine for the treatment of digestive upset. Grossman and Rasne revised the structure of sacacarin (150) by the total synthesis of putative sacacarin (149) and its lactone isomer 150 due to discrepancy of nuclear magnetic resonance (NMR) data between synthetic 149 and the natural product (Scheme 15).28 The synthesis was carried out by double Michael reaction leading to a 6-membered compound 143 and subsequent intramolecular Dieckmann reaction leading to decalone 145. After α-alkylation of Et2C=C(CN)2140 with dibromide 139, the remaining secondary bromide was replaced with diethyl malonate. Ozonolysis followed by alcoholysis to remove the propionyl group gave compound 141. Reaction with 4-trimethylsilyl-3-butyn-2-one 142 in the presence of KF promoted the double Michael reaction to give the 6-membered compound 143 in a highly diastereomeric ratio of 60:1. Treatment of 143 with HCl gas in anhydrous ethanol transformed the equatorial cyano group into an ethoxycarbonyl group in 5.6:1 diastereoselectivity by the Pinner reaction to afford keto-ester 144. Dieckmann condensation of 144 and subsequent enol etherification provided trans-decalone 145. After reaction with an equimolar amount of MeLi, the resulting tertiary alcohol was protected as a trimethylsilyl ether. Reduction of the ester with LiAlH4, followed by treatment with acid, furnished compound 146 having the requisite δ-lactone and enone moieties of putative sacacain 149. A furan moiety was introduced by the Wittig reaction of ylide 147. Attempts at catalytic hydrogenation of the double bond resulted mainly in over reduction of the furan ring, and the NMR data of the minor constituent 149 was not identical with that of the natural product. Then, the enone moiety of compound 149 was protected as a ketal and the lactone moiety was reduced to give the diol. The NMR data of the lactone 150 obtained by oxidation was identical with that of the natural product, thereby revising the structure of sacacarin (150).
Total syntheses of putative 149 and actual (±)-sacacarin (150).28
Clerocidin (156), an ent-neoclerodane having a unique oxygenated side chain, was isolated from Oidiodendron truncatum. The compound inhibits a broad spectrum of Gram-positive and Gram-negative bacteria in vitro and is active against P-388 lymphocytic leukemia in mice. Marko et al reported the synthesis of decalone 155 bearing the requisite functional groups for clerodane natural product syntheses by intramolecular reductive alkylation of the Morita–Baylis–Hillman (M-B-H) adduct 153 between aldehyde 152 and 2-cyclohexen-1-one (Scheme 16).29 The adduct 153 was obtained in good yield employing (n-Bu)3P and BINOL, the reaction conditions developed by Ikegami et al,30 which solved the issue encountered in the reactions of cyclic enones. Treatment of the acetate of 153 with excess Li in liquid ammonia generated ketyl radicals, in which coupling of the radicals, followed by the elimination of the acetoxy group and then conjugate reduction of the resulting enone proceeded sequentially to afford decalones 154 and 155.
Syntheses of clerodane 154 and 9-epi-clerodane 155 cores toward clerocidin (156).29
Terpentecin (169), also an ent-neoclerodane, was isolated from the culture broth of Kitasatosporia strain MF730-N6. Compound 169 inhibited the growth of Gram-positive and Gram-negative bacteria, and prolonged the survival period of mice bearing leukemia L-1210, P388 and Ehrlich ascites carcinoma, which is postulated to induce topoisomerase II-mediated DNA damage leading to cytotoxicity.
Theodorakis et al reported the synthesis of the terpentecin framework 168 (Scheme 17).9 Starting from Wieland–Miescher ketone analog 157,11 enone 160 was obtained by transformation of the allyl group, followed by selenylation and oxidation. Reduction with LiAlH4 provided β-alcohol at C7 selectively, which was transformed into epoxy-ketone 161. The epoxide opening by PhSeNa enabled introduction of the requisite β-hydroxyl group at C6 to give hydroxy-ketone 162. Though the Wittig reaction of 162 resulted in recovery, the Peterson olefination was successful to give methylene derivative 163. Catalytic hydrogenation in the presence of Wilkinson catalyst proceeded from the same side as the C6 hydroxyl group to afford α-methyldecalone 164 in >20:1 ratio. No stereoselectivity was observed with the Pd/C-catalyzed hydrogenation at atmospheric pressure. After introduction of an exo-methylene group at C4, ketone 166 was obtained by oxidation and isomerization of the exo-methylene at C4 by I2 catalysis. A β-hydroxyl group at C7 was installed by Davis reagent 167.
The same authors also synthesized the diastereomer 175 of compound 168 at C7 and C8 in a similar manner (Scheme 18).9 Ozonolysis and subsequent reduction followed by deprotection and protection led to hydroxy-ketone 170. The double bond at C3 was introduced by Wittig methylenation followed by I2 catalyzed isomerization and oxidation to give ketone 171. Transformation to enone 172 and subsequent enone transposition afforded enone 173, which was reduced with Li in liquid ammonia to furnish a β-methyl group at C8 of ketone 174 in 10:1 diastereomeric ratio. Hydroxylation at C7 by the Davis regent 167 proceeded selectively from the α-face of the molecule to give compound 175.
Synthetic study toward terpentecin (169) diastereomer.9
Arenarone (182), arenanol (183), and aureol (186) are cis-homoclerodanes that were isolated from the Caribbean sponges, Smenospongia aurea and Verongula gigantean; the compounds exhibited selective cytotoxicity against A549 human non-small cell cancer cells and anti-influenza-A virus. Katoh et al reported syntheses of these cis-homoclerodanes starting from the known aldehyde 17731 (Scheme 19).10 After addition of an anisole moiety to the aldehyde 177, ketone 179 was obtained by hydrogenolysis of a benzylic hydroxyl group. Introduction of a methylene group at C4 was carried out by the Takai method employing CH2Br2/Zn/TiCl432 to give exo-methylene 180, whereas Wittig, Peterson, and Tebbe methylenation resulted in the recovery of the starting ketone 179. Deprotection of the methylether, followed by aerobic oxidation of the phenol moiety in the presence of salcomine furnished arenarone (182). Reduction of the quinone moiety of arenarone (182) with sodium hydrosulfite provided arenarol (183), which was transformed with BF3·OEt2 to aureol (186) by sequential rearrangement via carbocation intermediates 184 and 185 and subsequent ether ring formation.
Total syntheses of (+)-arenarone (182), (+)-arenanol (183), and (+)-aureol (186).10
(−)-Methyl barbascoate (196) was isolated from Croton californicus, the leaves of which were used traditionally as a pain reliever for rheumatism and to stun fish by fishermen. Its absolute stereochemistry was simply assigned by comparison with CD data of (−)-methyl hardwickiate. Hagiwara et al accomplished total synthesis of (−)-methyl barbascoate (196) starting from known decalone 18733 derived from Wieland–Miescher ketone analog 176 (Scheme 20).34 Aldehyde 190 was obtained via a series of transformations, namely Wittig reaction, hydroboration, oxidation, and isomerization. The formyl group was protected as an acetal. After deprotection and oxidation leading to aldehyde 191, addition of 3-lithiofuran at C12 proceeded non-stereoselectively under a variety of reaction conditions. (12R)-Adduct 193 was transformed to the δ-lactone 194 via hemiacetal. Palladium catalyzed CO insertion to enol triflate 195 furnished (−)-methyl barbascoate (196), thereby establishing the absolute stereochemistry.
Total synthesis of methyl (−)-barbascoate (196).34
Furthermore, the same authors reported the second-generation synthesis of (−)-methyl barbascoate (196) in shorter steps, in which two ester groups at C4 and C8 were introduced at the same time by CO insertion into bis-enol triflate 198 (Scheme 21).35 Addition of 3-lithiofuran to aldehyde 200 resulted in the formation of unnatural (12S)-adduct 201 as a major isomer, in spite of the expectation of chelation of titanium between two carbonyl groups. An attempt at Mitsunobu inversion at C12 resulted in the preferential formation of (12S)-lactone. Conjugate reduction of the double bond at C7 by SmI2 proceeded regio- and stereoselectively to afford compound 201, whose stereochemistry is explained by the axial protonation to the samarium enolate. Isomerization at C8 furnished (−)-methyl barbascoate (196) in seven overall steps from known diketone 197.
Second-generation synthesis of methyl (−)-barbascoate (196).35
Salvinorin A (206), isolated from the Mexican sage Salvia divinorum, is a unique and densely functionalized furo-lactone neo-clerodane diterpenoid with seven asymmetric centers and five different oxygen functionalities (Figure 3).36,37 Nine congeners, from salvinorins B to J, have been identified so far.
Salvinorin A(206)~salvinorin J (215).
Salvinorin A (206) has the strongest hallucinogenic activity among known compounds and was identified as a selective non-nitrogeneous agonist of a κ-opioid receptor. Because a different mechanism from either LSD or other nitrogeneous hallucinogenic compounds was anticipated for its physiological activities, salvinorin A (206) has attracted much attention from synthetic organic chemists and physiologists as a lead compound for diseases associated with disorders of the central nervous system, in which a number of analogs have been prepared by semi-synthesis from salvinorin A (206).38-41 Issues on salvinorin syntheses are how to control seven stereogenic centers and to introduce five oxygen functionalities at the proper stages. Easy isomerization at C8 under weakly basic condition makes the control of the stereochemistry especially difficult. At the same time, stereoselective introduction of the furan at C12 might be an additional issue, which was encountered in the synthesis methyl barbascoate (196).
The first synthetic approach was reported by Rook et al (Scheme 22).42 Furan 218 was obtained by the conjugate addition of (−)-ephedrine to pentyn-4-one 216, followed by acid catalyzed cyclization. The Diels–Alder reaction of the furan 218 with methyl acrylate, followed by hydrolysis, provided adducts 219 and 220 in 66% ee. When the Diels–Alder reaction of the (+)-pseudoephedrine adduct of 216 was carried out in CH2Cl2, undesired endo-adduct 219 predominated in 99% yield, with 88% ee. Opening of the ether ring of the exo-adduct 220 with BBr3 and subsequent quenching with collidine, followed by acetylation led to A-ring precursor 222. C-ring precursor 226 was prepared by the Reformatsky reaction employing Rieke zinc.
Perlmutter et al synthesized racemic tricyclic precursor 232 targeting the C20-nor-analog of salvinorin A (234) (Scheme 23).43 Racemic diene 228 was obtained by the ene–yne metathesis reaction according to the protocol of Snapper et al.44 The Diels–Alder reaction of 228 with methoxy-quinone 229 proceeded in the presence of TiCl4 in toluene, although the desired endo-selectivity was not realized. After isomerization of the ring juncture of the endo-syn adduct 230 into trans-decalone 231, reduction of the enone moiety with sodium dithionate provided the target compound 233 without losing the methoxy group.
Synthetic study toward C20-norsalvinorin A (234).43
Hanquet et al carried out the synthesis of a chiral precursor 232 according to a pathway similar to that of Perlmuter et al as a part of their structure–activity relationship study (Scheme 24).45 The Diels–Alder reaction between chiral quinone 235 and chiral diene 228 furnished single adduct 236 by double asymmetric induction having the same stereochemistry as salvinorin A (206), in spite of the diene 228 and the dienophile 235 being a mismatched pair. Desulfurization accompanied isomerization of the ring juncture to give optically active Perlmutter precursor 232 having the requisite absolute stereochemistry for salvinorin (206).
Synthetic study of chiral precursor 232 toward C20-norsalvinorin A (234).45
The first total synthesis of salvinorin A (206) was accomplished by Evans et al and highlighted by the transannular domino Michael reaction of the 14-membered lactone 249 as a key step to construct the decaline framework 250 having the requisite functionality and stereochemistry (Schemes 25 and 26).46
Syntheses of precursors 243 and 246 by Evans et al.46
Syntheses of two fragments, aldehyde 243 and vinyliodide 246, were achieved as described in Scheme 25. After Ni-BINAP catalyzed asymmetric introduction of a dimethoxymethyl group into thiazolinethione 237, Claisen condensation gave β-ketoester 238, which led to the tri-substituted olefin 239 and subsequently to the aldehyde by DIBAL-H reduction. The aldol reaction of the aldehyde with chiral thiazolinethione 240 gave adduct 241, which was transformed into propargyl alcohol 242 via sequential transformations of the ester to amide, cleavage of the terminal double bond and ephedrine-mediated asymmetric addition of zinc acetylide. Semi-hydrogenation of the acetylene, dihydroxylation of resulting olefin and subsequent cleavage led to one precursor aldehyde 243.
Alcohol 245 was obtained by (R)-B-Me-CBS catalyzed asymmetric reduction followed by isomerization of the triple bond into the terminal, the internal carboalumination of which provided another precursor, vinyliodide 246.
Addition of the anion generated from vinyliodide 246 to aldehyde 243 proceeded almost stereoselectively by chelation control to give alcohol 247 (Scheme 26). After transformation to seco-acid 248 by protection and deprotection, the 14-membered lactone 249 was obtained according to the Shiina protocol.47
The first total synthesis of (−)-salvinorin A (206) by Evans et al.—Part 2.46
Treatment with TBAF at low temperature promoted the transannular domino Michael reaction to provide tricyclic compound 250 as a single diastereomer in high yield controlling the stereochemistry at C5, 9 and 10, which bore five requisite stereogenic centers of salvinorin A (206). Especially, it is notable that the correct stereochemistry at C12 was obtained, which has been difficult so far by the addition of 3-lithiofuran. Transformation to enol triflate at C7, followed by Pd catalyzed transfer hydrogenation furnished unsaturated δ-lactone, whose double bond at C7 was further reduced with L-Selectride stereoselectively to give the 8α-compound 251 as a result of kinetically and thermodynamically controlled axial protonation to enolate. Oxidation of the acetal into ester, isomerization at C8 and acetylation at C2 completed the total synthesis of (−)-salvinorin A (206) in a stereoselective and convergent manner in 29 overall steps.
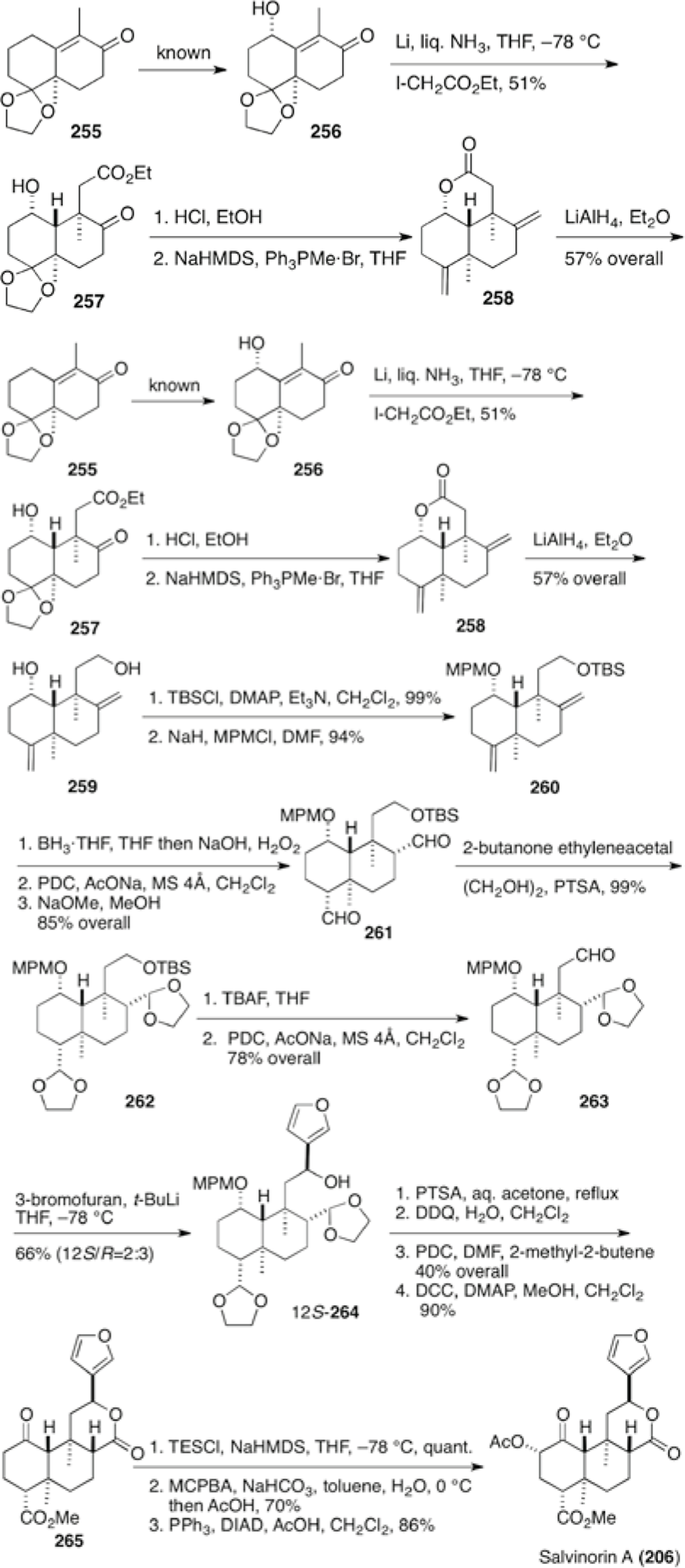
Hagiwara et al reported the second total synthesis of (−)-salvinorin A (206) in 20 overall steps based on their previous synthesis of methyl barbascoate (196) (Schemes 20 and 21), in which C12 was antipodal to salvinorin A (206). The synthesis was carried out starting from the known acetal 25648,49 derived from chiral Wieland-Miescher ketone analog 255 (Scheme 27),50 in which the stereochemical course of the synthesis was controlled explicitly. After reductive alkylation of 256 with lithium in liquid NH3, the double Wittig methylenation provided bis-methylene-lactone 258, whereas reaction with methoxymethylene triphenylphosphorane recovered the starting material. Selective protections of diol 259, hydroboration of methylenes and oxidation led to the bis-aldehyde. Equilibration with base gave bis-aldehyde 261 having the requisite stereochemistry at C4 and C8, which was protected as bis-acetal 262. The addition of 3-lithiofuran to aldehyde 263 proceeded non-stereoselectively. An attempt at Mitsunobu inversion of (12R)-alcohol 264 resulted in a diastereomeric mixture, probably due to the intervention of the SN1 pathway. An attempt at inversion at C12 by oxidation and reduction even by CBS catalysis also provided a diastereomeric mixture. After transformation of (12S)-alcohol 264 to hemi-acetal by deprotection followed by oxidation into keto-lactone, keto-ester 265 was obtained by esterification with DCC/DMAP. Introduction of the hydroxyl group at C2 was carried out by Rubottom oxidation to give the 2β-alcohol stereoselectively, which was inverted to the 2α-alcohol by the Mitsunobu reaction. Final acetylation furnished (−)-salvinorin A (206).
The second total synthesis of (−)-salvinorin A (206) by Hagiwara et al.50
Subsequently, the same authors described their second-generation synthesis of (−)-salvinorin A (206) starting from the hydroxy-ester 257, which was employed in their previous synthesis (Scheme 28).51 Major advances in the new pathway are the double introduction of two carbonyl groups at C4 and C8 and the stereocontrol at C12, thereby reducing synthetic steps and improving overall yield. After bis-triflate 267 from diketone 266 was transformed to Weinreb amide 268, addition of 3-lithiofuran provided furylketone 269, the palladium-catalyzed double CO insertion of which enabled the introduction of two unsaturated esters leading to bis-ester 246. Control of the stereochemistry at C12 was successfully accomplished by reduction with K-Selectride, which proceeded from the Si face of the carbonyl group selectively to result in (12S)-lactone 271 due to chelation of K-Selectride between two carbonyl groups. The double bond at C7 remained intact in the reduction. Subsequently, treatment of 271 with SmI2 resulted in the reduction of two double bonds at C3 and C7 to provide 4S,8S-lactone 272 as a major isomer as a result of the axial protonation to the bis-enolate generated by the double conjugate reduction, which was isomerized with base to the desired 4S,8R-273. The advanced intermediate 265 (Scheme 27) for (−)-salvinorin A (206) synthesis was obtained by Dess–Martin oxidation.
Hagiwara’s second-generation synthesis of (−)-salvinorin A (206).51
Forsyth et al elaborated the third total synthesis of salvinorin A (206) in 17 steps via construction of decalone 279 bearing three requisite stereogenic centers by exo-selective intramolecular Diels–Alder reaction followed by the introduction of the side chain by intramolecular Tsuji allylation52 leading to 280 (Scheme 29).53 Alcohol 276 was obtained through alkylation of dithioacetal 274 with iodide 275 derived from tartaric acid. Enol carbonate 278 was obtained after transformation of 276 to unsaturated ketone 277 via Horner–Wadsworth-Emmons reaction. The intramolecular Diels–Alder reaction of the enol carbonate 278 proceeded in high yield and stereoselectivity to give trans-decalone 279 after deprotection of the dithioacetal moiety. The intramolecular Tsuji allylation of the resulting carbonate 279 proceeded from the sterically less demanding β-face of the molecule to provide 9-allyl derivative 280. Transformations leading to salvinorin A (206) were carried out mainly according to the protocol of Hagiwara et al (Scheme 28).51 Palladium-catalyzed double CO insertion of bis-enol triflate provided bis-unsaturated ester 281. Cleavage of the terminal double bond of 281 led to the C12 aldehyde. The addition of 3-lithiofuran to the aldehyde in the presence of (R)-BINOL-Ti(Oi-Pr)4 furnished 12S-lactone 282 as a major isomer, which was difficult to control so far. Double conjugate reduction by SmI2 followed by deprotection afforded diols 283 and 284. Acetylation of the minor 8 R-284 occurred at C2 regioselectively and the oxidation furnished (−)-salvinorin A (206).
The third total synthesis of (−)-salvinorin A (206) by Forsyth et al.53
Salvinorin F (211), though a minor constituent in nature, is anticipated to be a common synthetic intermediate for the transformation into salvinorins C to J, all of which have Δ3-unsaturated esters at C4. Hagiwara et al synthesized salvinorin F (211) from the lactone 271, which was employed in the previous synthesis of salvinorin A (206) (Scheme 30).54 Treatment of the lactone 271 with SmI2 in the presence of excess pivalic acid resulted in regioselective reduction of the double bond at C7 to give 8α- and 8β-lactone in a 3:1 diastereomeric ratio. Deprotection of the 8β-isomer 286 led to salvinorin F (211), along with isomerization of the 8α-isomer 287.
In this article, total syntheses of clerodanes reported from 2000 to 2017 have been reviewed. In spite of the plethora of clorodane diterpenoids, examples of total syntheses are limited. Among the successful ones, there still remain some issues to improve lengthy procedures along with low stereoselectivity, especially in the introduction of a pendant furan ring. Total syntheses of clerodanes having a hydro-furofuran-based side chain are future challenges, as are those of ring-seco, degraded and rearranged clerodanes. Discovery of new clerodane diterpenoids is still continuing. Abundance of clerodanes might increase new fascinating biological and pharmacological activities. Furthermore, these aspects will facilitate the study of total syntheses of clerodanes in which the appearance of novel strategy and reactions is expected.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
HaradaNUdaH. Absolute stereochemistries of 3-epicaryoptin, caryoptin, and clerodin as determined by chiroptical methods. J Am Chem Soc. 1978;100(25):8022-8024.
3.
TrivediGKomuraHKuboINakanishiKJoshiBS. The ent-neo-clerodane absolute configuration of ajugarins. J Chem Soc Chem Commun. 1979;20(20):885-886.
4.
KuboIKidoMFukuyamaY. X-Ray crystal structure of 12-bromoajugarin-I and conclusion on the absolute configuration of ajugarins. J Chem Soc Chem Commun. 1980;19(19):897-898.
5.
RogersDÜnalGGWilliamsDJ, et al. The crystal structure of 3-epicaryoptin and the reversal of the currently accepted absolute configuration of clerodin. J Chem Soc, Chem Commun. 1979;3(3):97-99.
6.
OhsakiA. Structural diversity and bioactivity of labdane and clerodane diterpenoids. J. Syn. Org. Chem., Jpn..2004;62(9):872-881.
7.
TokoroyamaT. Synthesis of clerodane diterpenoids and related compounds-stereoselective construction of the decalin skeleton with multiple contiguous stereogenic centers. Synthesis. 2000;5:611-633.
8.
HagiwaraH UUdaH. Synthesis of optically pure (4aS)-(+)- or (4aR)-(-)-1,4a-dimethyl-4,4a,7,8-tetrahydronaphthalene-2,5(3H,6H)-dione and the use for application to the synthesis of an inhibitor of steroid biosynthesis. J. Org. Chem.. 1988;53(10):2308-2311.
9.
LingTRivasFTheodorakisEA. Stereoselective synthesis of the fully functionalized core fragment of terpentecin. Tetrahedron Lett. 2002;43(50):9019-9022.
10.
NakataniMNakamuraMSuzukiAFuchikamiTInoueMKatohT. Enantioselective total synthesis of (+)-aureol via a BF3 -Et2O promoted rearrangement/cyclization reaction of (+)-arenarol. ARKIVOC. 2003;8:45-57.
11.
MerrittATPouwerRHWilliamsDJWilliamsCMLeySV. The clerodane ring system: investigating the viability of a direct Diels-Alder approach. Org Biomol Chem. 2011;9(13):4745-4747.
12.
LiuH-JZhuJ-LChenI-CJankowskaRHanYShiaK-S. The total synthesis of racemic teucvin and 12-epi-teucvin. Angew. Chem. Int. Ed..2003;42(16):1851-1853.
13.
ChenI-CWuY-KLiuH-JZhuJ-L. Total syntheses of (±)-montanin A and (±)-teuscorolide. J Chem Soc Chem Commun. 2008;50(39):4720-4722.
14.
Y-KWChenI-CZhuJ-LLiuH-J. Efficient installation and elaboration of C4-C6 fused furan moiety in the total synthesis of Teucrium clerodane diterpenoids. Heterocycles. 2014;89(9):2091-2104.
15.
LiuXLeeC-S. Total synthesis of (−)-teucvidin. Org Lett. 2012;14(11):2886-2889.
16.
ArnsSBarriaultL. Concise synthesis of the neo -clerodane skeleton of teucrolivin A using a pericyclic reaction cascade. J Org Chem. 2006;71(5):1809-1816.
17.
YeQQuPSnyderSA. Total syntheses of scaparvins B, C, and D enabled by a key C-H functionalization. J Am Chem Soc. 2017;139(51):18428-18431.
18.
LiuHJYLH, JDW, ShiaKS. An efficacious synthetic strategy for cis-clerodane diterpenoids. Application to the total synthesis of (±)-6β-acetoxy-2-oxokolavenool. Synlett. 2001;11:1805-1807.
19.
ChenMSWhiteMC. A predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. Science. 2007;318(5851):783-787.
20.
BigiMAReedSAWhiteMC. A predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. J Am Chem Soc. 2012;134(23):9721-9726.
21.
LiuHJShiaKS. Synthetic studies on clerodane diterpenoids. 4. The total synthesis of (±)-6β-acetoxy-2-oxokolavenool. Tetrahedron. 1998;54(44):13449-13458.
22.
MihelichEDEickhoffDJ.EickhoffD. A one-pot conversion of olefins to alpha, beta-unsaturated carbonyl compounds. An easy synthesis of 2-cyclopentenone and related compounds. J Org Chem. 1983;48(22):4135-4137.
23.
WuJ-DShiaKSLiuHJ. An improved general synthetic approach to cis-clerodane diterpenoids. A more efficient total synthesis of (±)-6-acetoxy-2-oxokolavenool. Tetrahedron Lett. 2001;42(25):4207-4209.
24.
KatoMKosugiHIchiyanagiTet al. New entry to the synthesis of clerodane diterpenens. The first enantioselective syntheses of 7-oxo-kolavenic acid and methyl solidagonate. Tetrahedron. 2000;57(39):8243-8256.
25.
MüllerDSUntiedtNLDieskauALacknerGLOvermanLE. Constructing quaternary stereogenic centers using tertiary organocuprates and tertiary radicals. Total synthesis of trans-clerodane natural products[erratum appears in J Am Chem Soc. 2015;137(14):4874]. J Am Chem Soc. 2015;137(2):660-663.
26.
PiersEWaiJSM. Stereoselective total synthesis of the marine sesterterpenoid (±)-palauolide. Can J Chem. 1994;72(1):146-157.
27.
SlutskyyYJamisonCRLacknerGL, et al. Short enantioselective total syntheses of trans-Clerodane diterpenoids: convergent fragment coupling using a trans-decalin tertiary radical generated from a tertiary alcohol precursor. J Org Chem. 2016;81(16):7029-7035.
28.
GrossmanRBRasneRM. Short total syntheses of both the putative and actual structures of the clerodane diterpenoid (+/-)-sacacarin by double annulation. Org Lett. 2001;3(25):4027-4030.
29.
WasnairePWiauxMTouillauxRMarkóIE. Reductive cyclisation of Morita–Baylis–Hillman adducts. A simple approach towards substituted hydrindanones and decalones. Tetrahedron Lett. 2006;47(6):985-989.
30.
YamadaYMAIkegamiS. Efficient Baylis–Hillman reactions promoted by mild cooperative catalysts and their application to catalytic asymmetric synthesis. Tetrahedron Lett. 2000;41(13):2165-2169.
31.
KatohTNakataniMShikitaS, et al. Studies toward the total synthesis of popolohuanone E: enantioselective synthesis of 8-O-methylpopolohuanone E. Org Lett. 2001;3(17):2701-2704.
32.
TakaiKHottaYOshimaKNozakiH. Wittig-type reaction of dimetallated carbodianion species as produced by zinc reduction of gem-polyhalogen compounds in the presence of Lewis acids. Bull Chem Soc Jpn. 1980;53(6):1698-1702.
33.
HagiwaraHInomeKUdaH. A total synthesis of antibacterial clerodane, 16-hydroxycleroda-3,13(14)Z-dien-15,16-olide. J Chem Soc Perkin Trans. 1995;1(7):757-764.
34.
HagiwaraHHamanoKNozawaMHoshiTSuzukiTKidoF. The first total synthesis of (-)-methyl barbascoate. J Org Chem. 2005;70(6):2250-2255.
35.
HagiwaraHHonmaNKinugawaKSatoSHoshiTSuzukiT. Second generation synthesis of the neo-clerodane diterpenoid methyl barbascoate. Nat Prod Commun. 2013;8(7):873-875.
36.
PrisinzanoTE. Natural products as tools for neuroscience: discovery and development of novel agents to treat drug abuse. J Nat Prod. 2009;72(3):581-587.
37.
CasselmanINockCJWohlmuthHWeatherbyRPHeinrichM. From local to global-fifty years of research on Salvia divinorum. J Ethnopharmacol. 2014;151(2):768-783.
38.
PrisinzanoTERothmanRB. Salvinorin A analogs as probes in opioid pharmacology. Chem Rev. 2008;108(5):1732-1743.
39.
LozamaAPrisinzanoTE. Chemical methods for the synthesis and modification of neoclerodane diterpenes. Bioorg Med Chem Lett. 2009;19(18):5490-5495.
40.
RileyAPGroerCEYoungDEwaldAWKivellBMPrisinzanoTE. Synthesis and κ-opioid receptor activity of furan-substituted salvinorin A analogues. J Med Chem. 2014;57(24):10464-10475.
41.
RoachJJShenviRA. A review of salvinorin analogs and their kappa-opioid receptor activity. Bioorg Med Chem Lett. 2018;28(9):1436-1445.
42.
LinghamARHügelHMRookTJ. Studies towards the synthesis of salvinorin A. Aust J Chem. 2006;59(5):340-348.
43.
BergmanYEMulderRPerlmutterP. Total synthesis of 20-norsalvinorin A. 1. Preparation of a key intermediate. J Org Chem. 2009;74(6):2589-2591.
44.
CheungAKMurelliRSnapperML. Total syntheses of (+)- and (-)-cacospongionolide B, cacospongionolide e, and related analogues. Preliminary study of structural features required for phospholipase a2 inhibition. J Org Chem. 2004;69(17):5712-5719.
45.
LanfranchiDABourCHanquetG. Enantioselective access to key intermediates for salvinorin A and analogues. European J Org Chem. 2011;2011(15):2818-2826.
46.
ScheererJRLawrenceJFWangGCEvansDA. Asymmetric synthesis of salvinorin A, a potent κ opioid receptor agonist. J Am Chem Soc. 2007;129(29):8968-8969.
47.
ShiinaIKubotaMOshiumiHHashizumeM. An effective use of benzoic anhydride and its derivatives for the synthesis of carboxylic esters and lactones: a powerful and convenient mixed anhydride method promoted by basic catalysts. J Org Chem. 2004;69(6):1822-1830.
48.
ShimizuTHiranumaSYoshiokaH. Regio- and stereoselective functionalizations of the Wieland-Miescher ketone derivatives at the C-8 position. Chem Pharm Bull. 1989;37(7):1963-1965.
49.
AladroFJGuerraFMMoreno-DoradoFJBustamanteJMJorge Zacarı́asDMassanetGM. Enantioselective synthesis of (+)-decipienin A. Tetrahedron. 2001;57(11):2171-2178.
50.
NozawaMSukaYHoshiTSuzukiTHagiwaraH. Total synthesis of the hallucinogenic neoclerodane diterpenoid salvinorin A. Org Lett. 2008;10(7):1365-1368.
51.
HagiwaraHSukaYNojimaTHoshiTSuzukiT. Second-generation synthesis of salvinorin A. Tetrahedron. 2009;65(25):4820-4825.
52.
ShimizuIOhashiYTsujiJ. A new one-pot method for α,α-diallylation of ketones based on the palladium-catalyzed reaction of allylic carbonates and allyl β-keto carboxylates under neutral conditions. Tetrahedron Lett. 1983;24(36):3865-3868.
53.
LineNJBurnsACButlerSCCasbohmJForsythCJ. Total Synthesis of (−)-Salvinorin A. Chem. Eur. J.. 2016;22(50):17983-17986.
54.
HagiwaraHNojimaTSukaYHoshiTSuzukiT. First total synthesis of the neo-clerodane diterpenoid salvinorin F. Nat. Prod. Commun. 2011;6(3):333-335.