
Abstract
Methanol extracts of the rhizomes or fruit of Amomum celsum Lamxay & M.F. Newman (Zingiberaceae) yielded 5 compounds one of which, 1,3S,5S-trihydroxy-1-(4-hydroxyphenylethyl)cyclohexane (
The nontimber genus Amomum (Zingiberaceae) was first described by Linnaeus in 1747. Today, some of approximately 170 Amomum species such as A. tsao-ko Crevost & Lemarié, A. villosum Lour., A. krervanh PierreexGagnep., and A. xanthioides Wall. have been recognized as plants of economic importance because of their extensive use as spices and medicinal herbs. The number of phytochemical studies of Amomum species is relatively small including A. pavieanum,
1
A. krervanh,
2
A. koenigii,
3
A. aculeatum,
4–7
A. xanthioides,
8
A. subulatum,
9–11
A. tsao-ko,
12–14
A. muricarpum,
15,16
and A. biflorum.
17
Amomum celsum has an area of occurrence of 314 km2 from Kon Tum province in Vietnam and Attapeu province in Laos.
18
There has been no phytochemical study of A. celsum. We prepared water-soluble fractions from the rhizomes or fruit of A. celsum in Vietnam and isolated compounds
MeOH extracts were prepared individually from the dried rhizomes or fresh fruit of A. celsum by extraction at room temperature. Two water-soluble fractions were prepared by successive fractionation of the MeOH extracts with n-hexane, dichloromethane, and ethyl acetate. The water-soluble fractions were chromatographed successively on highly porous Diaion HP-20, RP-18, and silica gel to afford 5 compounds. The structures of the known compounds
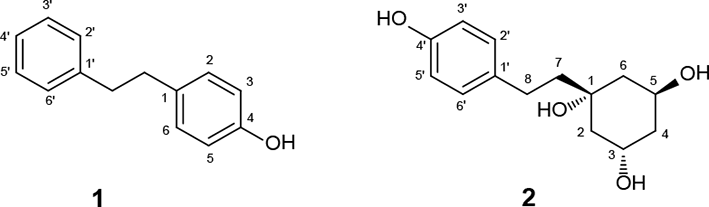
Structures of 1 and 2.
Compound
Compound

1H-1H COSY, HMBC, and NOESY correlations of 2.
The mutiplicities and values of coupling constants of the cyclohexyl protons were consistent with the assignments. Judging from the stability of cyclohexane conformations, a chair conformation that is stabilized by an intramolecular hydrogen bond between 2 diaxial hydroxy groups at C-1 and C-3 was chosen. Thus, the values of diaxial coupling constants of H-6ax at δ H 1.42 (J = 10.5 Hz) and H-4ax at δ H 1.47 (J = 11.5 Hz) indicated that H-5 (δ H 4.23, m) was also axially orientated. Both axial protons coupled with H-4eq (δ H 2.15) and H-6eq (δ H 2.08) by the same geminal coupling constant (J = 13.0 Hz). The values of the coupling constants measured for H-2ax at δ H 1.58 (J = 3.0 Hz) and H-4ax at δ H 1.47 (J = 2.0 Hz) indicated the equatorial orientation of H-3 (δ H 4.26, m). Long-range W-couplings (4 J = 1.7 Hz) were observed between H-2eq (δ H 1.87) and H-6eq and H-4eq and H-6eq. The nuclear Overhauser effect spectroscopy (NOESY) experiments (Figure 2) were not selective to rule out 1H-1H COSY signals for H-3 and H-5; however, the NOESY correlations between H2-7 and H-2ax and H-6ax confirmed the equatorial orientation of the phenethyl group, thus confirming the axial orientation of the hydroxy group at C-1.
The relative structure assembled by the spectroscopic data was confirmed by the crystallographic analysis and the absolute structure is as shown in Figure 3, where the Flack paramter was 0.02(4). Two independent molecules and water molecules are included in an asymmetric unit, and one water molecule forms a hydrogen bond with 2 independent molecules. The absolute configuration was further confirmed by the modified Mosher’s method
24
as shown in Figure 4. On the basis of the extensive NMR and X-ray analysis, the structure of
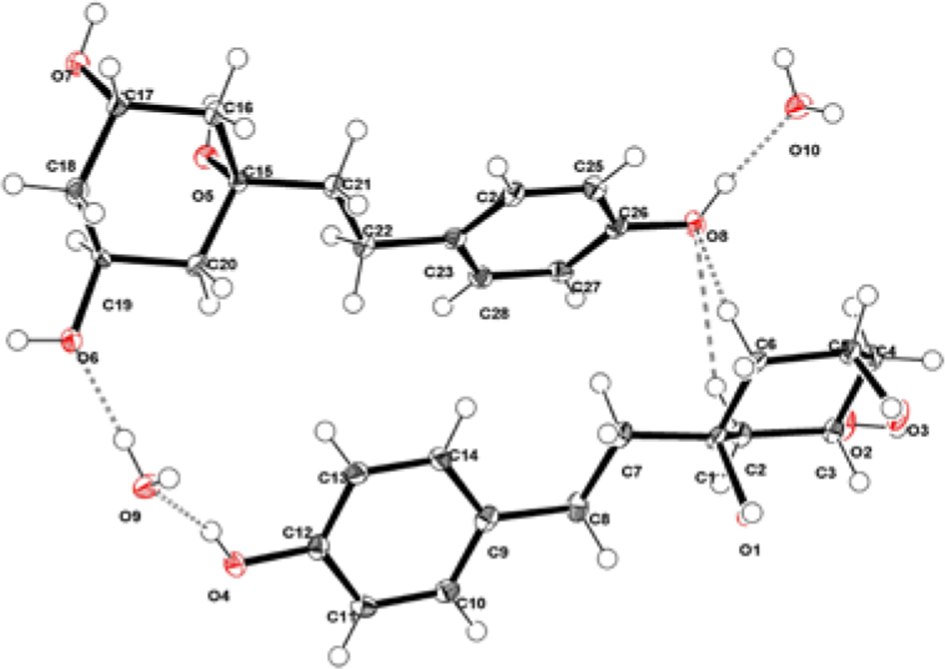
X-ray crystallographic structure (H bonding) of 2.

Mosher’s esters of 2. Δδ values are in ppm (δS–δR ).
Compound

Plausible biosynthetic pathway of 1 and 2.
Experimental
General Procedure
Optical rotations were measured on a JASCO P-2200 digital polarimeter. IR spectra were recorded on a Jasco FT-IR 6100 spectrometer. ESI-MS spectra were obtained using a LC-MS 6310 Agilen Ion Trap system. HR-ESI-MS spectra were measured on a Thermo Fischer Scientific LTQ Orbitrap XL mass spectrometer. 1H, 13C NMR, DEPT, 1H-1H COSY, HSQC, HMBC, and NOESY spectra were recorded on a Bruker Avance 500 NMR or 600 MHz spectrometers. The diffraction experiment was performed with a Bruker D8VENTURE system (PHOTON-100 CMOS detector, CuKα: λ = 1.54178 Å). All nonhydrogen atoms were refined anisotropically. The hydrogen atoms were refined isotropically on the calculated positions using a riding model except for hydroxy hydrogen. Absorption correction was performed by an empirical method implemented in SADABS. Structure solution and refinement were performed using SHELXT-2014/5 and SHELXL-2018/3. Silica gel (Merck, Germany) and Diaion HP-20 (Mitsubishi, Japan) were used for column chromatography (CC). Merck LiChrolut® (reversed phase)RP-18 (Merck, Germany) cartridges were used for solid-phase extraction (SPE). Thin-layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates (Merck, Germany).
Plant Material
Fresh rhizomes and fruit of A. celsum Lamxay & M.F. Newman were collected in Kon Tum province, Vietnam, in July 2016. The plant material was identified by one of the authors, Dr Nguyen Quoc Binh (Vietnam National Museum of Nature, Vietnam Academy of Science and Technology, Hanoi, Vietnam). A voucher sample (No. EF-12-15) has been deposited at the same Museum.
Extraction and Isolation
The fresh rhizomes and fruit were cleaned and air-dried to remove surface water in the shade. The rhizomes were dried in an oven at 40°C to 50°C then powdered. The fruit (3 kg) was crushed in an electric blender. The dried powdered rhizomes (2 kg) or fruit paste of A. celsum were extracted with MeOH at room temperature 3 times, each time for 7 days. After filtration, the MeOH extracts were combined separately and evaporated under reduced pressure to afford 2 corresponding MeOH extracts which were suspended in distilled water. The suspensions were partitioned between water and n-hexane, CH2Cl2, and EtOAc. The water phases were concentrated and chromatographed on a Diaion HP-20 column eluted with gradient mixtures of MeOH-H2O (20%, 40%, 60%) and MeOH yielding 4 corresponding fractions. From the fruit: The 100% MeOH fraction (89 mg) was separated by silica gel CC with gradient mixtures of n-hexane-EtOAc-HCO2H (10:3:1, 10:5:1, 10:7:1, 10:10:1) to give 2 fractions. Compound
4-Hydroxybibenzyl (1)
Amorphous powder.
IR (film) νmax 3368, 1600, 1514, 1452, 1251 cm−1.
1H NMR (500 MHz, CD3OD): 2.86 (4H, m, 2H α , 2H β ), 6.68 (2H, d, J = 8.5 Hz, H-2, H-6), 6.97 (2H, d, J = 8.5 Hz, H-3, H-5), 7.15 (3H, m), 7.24 (1H, t, J = 7.5 Hz), 7.22 (1H, t, J = 7.5 Hz) (5 H, H-2′, H-3′, H-4′, H-5′, H-6′).
13C NMR(125 MHz, CD3OD): 38.3 (C α ), 39.5 (C β ), 116.0 (C-2, C-6), 126.8 (C-4′), 129.2 (C-3′, C-5′), 129.6 (C-2′, C-6′), 130.4 (C-3, C-5), 133.9 (C-1), 143.3 (C-1′), 156.4 (C-4).
Negative-ion HR-ESI-MS: m/z [M–H]– calcd for C14H13O: 197.0961, found: 197.0972.
1,3S,5S-Trihydroxy-1-(4-Hydroxyphenylethyl)Cyclohexane (2)
Colorless plates.
MP:. 139°C-141°C.
IR (film) νmax 3335, 1559, 1513, 1419, 1237 cm−1.
1H NMR (500 MHz, CD3OD): 1.42 (1H, dd, J = 13.0 Hz, 10.5 Hz, H-6ax), 1.47 (1H, ddd, J = 13.0 Hz, 11.5 Hz, 2.0 Hz, H-4ax), 1.58 (1H, dd, J = 14.0 Hz, 3.0 Hz, H-2ax), 1.73 (2H, ddd, J = 9.5 Hz, 7.5 Hz, 2.0 Hz, 2 H-7), 1.87 (1H, dddd, J = 14.0 Hz, 3.6 Hz, 1.7 Hz, 1.7 Hz, H-2eq), 2.08 (1H, dddd, J = 13.0 Hz, 2.0 Hz, 2.0 Hz, 1.7 Hz, H-6eq), 2.15 (1H, br d, J = 13.0 Hz, H-4eq), 2.61 (2H, m, 2 H-8), 4.23 (1H, m, H-5), 4.26 (1H, m, H-3), 6.70 (2H, d, J = 8.5 Hz, H-3′, H-5′), 7.02 (2H, d, J = 8.5 Hz, H-2′, H-6′).
13C NMR (CD3OD): 29.5 (C-8), 41.5 (C-2), 42.5 (C-4), 46.7 (C-6), 46.9 (C-7), 64.4 (C-5), 69.1 (C-3), 75.6 (C-1), 116.1 (C-3′, C-5′), 130.2 (C-2′, C-6′), 134.8 (C-1′), 156.3 (C-4′).
Positive-ion HR-ESI-MS: m/z [M + Na]+ calcd for C14H20O4Na: 275.1254, found: 275.1252.
Preparation of (S)- and (R)-MTPA Diesters (2a and 2b) From 2
A solution of
(S)-MTPA Diester (2a)
1H NMR (600 MHz, CDCl3): 1.5148 (1H, m, H-2ax), 1.572 (1H, m, H-6ax), 1.5580 (1H, m, H-4ax), 1.7852 (2H, m, 2 H-7), 2.0478 (1H, m, H-2eq), 2.2868 (1H, br d, J = 13.0 Hz, H-6eq), 2.3648 (1H, br d, J = 13.0 Hz, H-4eq), 2.7509 (2H, m, 2 H-8), 3.5486 (3H, s, -OCH3), 3.6834 (3H, s, -OCH3), 4.3790 (1H, m, H-3), 5.6981 (1H, m, H-5), 7.04 (2H, d, J = 8.5 Hz, H-3′, H-5′), 7.22 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.40–7.65 (10H, m, aromatic protons).
Positive-ion HR-ESI-MS: m/z [M + Na]+ calcd for C34H34O8F6Na: 707.2050, found: 707.2052.
(R)-MTPA Diester (2b)
1H NMR (600 MHz, CDCl3): 1.4925 (1H, t, J = 12.0 Hz, H-6ax), 1.5198 (1H, m, H-2ax), 1.6581 (1H, m, H-4ax), 1.7669 (2H, m, 2 H-7), 2.0546 (1H, br d, J = 13.0 Hz, H-2eq), 2.2378 (1H, br d, J = 13.0 Hz, H-6eq), 2.4265 (1H, br d, J = 13.0 Hz, H-4eq), 2.7307 (2H, m, 2 H-8), 3.5513 (3H, s, -OCH3), 3.6821 (3H, s, -OCH3), 4.4001 (1H, m, H-3), 5.6988 (1H, m, H-5), 7.031 (2H, d, J = 8.5 Hz, H-3′, H-5′), 7.211 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.40-7.65 (10H, m, aromatic protons).
Positive-ion HR-ESI-MS: m/z [M + Na]+ calcd. for C34H34O8F6Na: 707.2050, found: 707.2048.
X-Ray Crystallographic Analysis of 2
C14H22O5, M r = 270.31. Monoclinic, space group P21, Z = 4, D calc = 1.321 g cm−3, a = 6.1296(14), b = 7.0606(16), c = 31.400(7) Å, β = 90.218(5)°, V = 1359.0(5) Å3, 16 182 observed and 5191 independent [I > 2σ(I)] reflections, 381 parameters, final R 1 = 0.0324, wR 2 = 0.1012, S = 1.095 [I > 2σ(I)]. Flack parameter: χ = 0.02(4). The largest difference peak and hole were 0.314 and –0.355 eÅ–3, respectively.
Supplementary X-ray crystallographic data for
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2017.41.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.