
Abstract
Diabetes technology is a dynamically evolving field. Sometimes the pace of evaluation of new diabetes technologies does not keep pace with its dynamic development. This leads to a dilemma: either the evaluation lags behind the developing technologies or diabetes technologies are used without sufficient evaluation. This situation is known as the Catch 22 dilemma. The aim of this paper is a discussion of ideas for a timely assessment, taking account of the speed of technological development and the need for evidence and safety improvement.
Introduction
Diabetes technology is a very dynamic field that is providing new solutions for the care of people with diabetes. The question of what evidence is required before new or updated diabetes technology solutions can be brought to market and used arises for manufacturers, reimbursement or regulatory agencies, and users of these technologies. Evidence can be generated by randomized clinical efficacy trials (RCTs), which show the superiority or inferiority of a newly developed diabetes technology over existing standards, or by real-world data (RWD), which are data collected in routine care that show the effectiveness of a new diabetes technology in diabetes care. Given the dynamic development of diabetes technologies, it is not uncommon to find that the pace of evidence generation is too slow for the full benefits of these new technologies to be realized in diabetes care. With this editorial, we would like to stimulate a discussion on the level of evidence and safety analysis that is required for the timely availability of new diabetes technologies for the care of people with diabetes.
The scope of the problem
With each new product development, the question is, do people with diabetes (PwD) truly benefit from it? This not only holds true for new anti-hyperglycemic drugs, but also for new medical products used for diabetes therapy, might they be for diagnostic purposes, like systems for continuous glucose monitoring (CGM), 1 or insulin administration, like pumps 2 or pens 3 . In our digital age, software used by PwD themselves, like Apps on their smartphones, or more complex software programs used for virtual diabetes treatment, also must demonstrate that their use is worth the investment. Evidence regarding “efficacy” is a term used by regulatory agencies when it comes to the approval of new products for market entrance. Payers, that is, health care insurance companies (or related agencies), are interested in the effectiveness of their decision regarding the reimbursement of a new drug or technical device, which means how well a product performs in the real world. Efficacy is how well a medical product performs in a highly controlled, but somewhat unrealistic environment that is part of a clinical trial for a limited type of of patients. Efficacy is effectiveness measured in the best possible conditions. 4
It seems obvious that the performance of a product in a setting of a clinical trial with an adequate study design is linked to every new product intended for treating PwD. When it comes to the clinical development of new drugs, the performance of clinical trials in 3 development phases is mandatory for regulatory approval. This is a complex process that takes several years and is combined also with a huge financial investment. However, once a given drug is approved, the pharmaceutical companies have several years in which only they can sell the given product to counterbalance all these efforts before the loss of patent protection and competition from copycat products show up or better products come along to supplant this product in the marketplace.
The story is different when it comes to medical devices, especially in a world in which new technology is being developed with increasing speed from year to year. New generations of medical devices are frequently coming to the market because the progress made by a lot of research and development enables improvements of the products that are supposedly so relevant, that users can benefit from short product life cycles. However, this type of product cycle in turn generates a dilemma: is it neccessary to perform RCTs for each new generation of a product, which are the best way to generate high-level evidence? These trials are quite time-consuming and costly. From the decision to perform such a clinical study to the availability of the study results and publication of these results, it easily takes several years. In other words, once the evidence has been collected that documents the benefits of using a given next generation medical device, there is a high risk that this product is outdated or even not on the market anymore. Do the results obtained with 1 generation of a medical product, for example, a given CGM system, also hold true for the next generation of this product, which might have some significantly different features?
It is our impression that even though more medical devices (including software products) come to the diabetes market year by year, the number of good clinical studies performed with these to generate high-level evidence is not increasing, but decreasing. In addition, many clinical trials with new medical products are of insufficient study duration and are based on a sample size studied that is too small. Quite often in the respective publication, no information is given at all about the effect size that can be detected with the number of patients included. Another problem is that with small incremental improvements in devices, and therefore with small increments in improved outcomes, the number of subjects necessary for a well-powered study to show a statistically significant improvement can be quite large. For that reason, some studies of next-generation products have limited their outcomes to safety, with the implication being that if the system is safe, then it is not necessary to show statistically significant improved outcomes. The major reason for this dearth of high-quality trials of next-generation slightly upgraded devices with a short turnaround from generation to generation is the cost of clinical trials. At the same time, new Medical Device Regulations (MDR) are in place in Europe that demand the mandatory performance of clinical studies for the approval of medical products, which was always the case in the United States.
This editorial discusses several evidence generation issues that the technology community needs to agree on in order to reconcile the Catch 22 dilemma of providing the best safety and efficacy data while not slowing innovation in diabetes technology. Such an agreement could accelerate market access for new products, increase knowledge about efficacy and safety, and reduce unnecessary costs, making new products affordable for more people with diabetes.
Updates of Software and Hardware in Medical Products
Diabetes software is a computer program, which turns into a medical product when it is somehow involved in the therapy of PwD. Digital hardware is a medical device that performs a task or makes a measurement and is either guided by software or which communicates with software. A potential problem for software manufacturers is the need for frequent product upgrades. New versions of digital health tools might come along with improved features and performance; however, the option of constantly upgrading the product with the latest and best-selling technology (even in minor ways sometimes), represents a regulatory issue, especially concerning safety aspects. In this respect, software differs from hardware. The software can be upgraded within weeks or months if necessary, but hardware tends to have a life cycle of several years. Nevertheless, modern medical products are most often a combination of hardware and software. Also, hardware undergoes changes in various ways – also from a manufacturing point of view – and there is no fixed rule for how much of a given change warrants a new product name/generation with such substantial changes that a new evaluation of the clinical performance is justified/needed. Some manufacturers favor stability and do a lot of remodeling while keeping the product name the same and others want to frequently announce they are selling a “new and improved” product even if they are not doing much remodeling.
The problem of seeking evidence in the face of upgrades is also a problem for cyber security standards. Such standards are based on evidence of safety and even if the product changes “slightly,” then there might be concerns about continued security. It is difficult to motivate manufacturers to retest slightly modified products for security just as it is to retest them for clinical benefits. The U.S. Food and Drug Administration (FDA) as the regulatory authority regards a software change that is made to improve a security problem that was not recognized when the product was approved, as one that does not require a review by the agency. However, that policy does not apply to upgrades intended to improve clinical outcomes.
Is Evidence Truly Needed?
This might appear to be a nonsensical statement at first glance because without evidence no regular clearance can be achieved and also no reimbursement. However, some of the – digital – companies act aggressively (ie, disruptively) and bring products to the market (yes, these might not be as highly regulated as the medical world . . . ) even though these do not comply with certain laws and rules. If your pockets are deep enough to pay many lawyers for several years and the financial benefits from doing so are so big, that this counterbalances the costs for legal battles, then this might be an illegal and unethical approach that works.
In the last few years medical products used for the therapy of PwD were introduced successfully to the market in certain countries in an unusual way. Sometimes they are labeled as being for wellness rather than for a medical diagnosis. This approach might annoy physicians and pharmacists when these products are sold via the internet and not via conventional ways medical products were handled until recently; however, it might be that such approaches are appropriate in our digital world.
People with diabetes—like all of us—are used to having access to products 24/7 and don’t understand why this should be different for medical products that they need. So, the patients/users have built up pressure, also at the political level, to have access to such products (especially CGMs) even if they are not approved for some medical indications (because a national regulatory body has determined them to be low risk) or even if no reimbursement is provided. If the benefits of a given product are high enough, then PwD are willing to pay for these out of pocket. This has pushed not only the given product ahead but has also changed to a given extent the way products for PwD are introduced into the market. Some barriers are not as high and impregnatable as they might look at the first glance. For example, some insurance companies in Germany simply ignored their own rules for reimbursement of rtCGM systems—usually, they are quite strict in applying them—once they realized that customers might switch to a different company that is covering the costs for certain CGM systems. So, in case their business is under pressure, such insurance companies show more flexibility than one might expect.
Another topic related to a disconnect between evidence and adoption is if a given product is not approved in a given country but in another one, a PwD can simply order the item on the internet and has thereby access to it. Can you blame this person for doing so if they regard this as helpful/essential? Yes, there might be risks involved in doing so and there is probably no liability of the manufacturer if the PwD use such products obtained illegally; however, PwD have built systems for automated insulin delivery (AID) themselves (‘do it yourself’; DIY), that enable them to achieve quite good glucose control. An example is the Loop algorithm that recently received FDA clearance as an interoperable automated glycemic controller. The movement toward DIY AID systems is pushing the development of new products for the treatment of PwD ahead. The DIY community is also exhorting regulators to loosen the evidentiary requirements for the clearance of components of AID systems.
What are the lessons learned from such stories—also from an ethical point of view—when it comes to evidence and generation of evidence? Medical products used in diabetes therapy represent only a small portion of the huge world of medical products in general; however, probably the developments seen in this space can push ahead the necessary changes (see below) in the way medical products are approved and reimbursed. The digital revolution in the last 20 years does not stop when it comes to medical products. This, in turn, has also consequences when it comes to the generation of evidence on what is truly needed for what, it appears as if the current approach needs to keep up with the times.
Evidence for Which Purpose?
Regulatory agencies like the FDA ask for an improvement in HbA1c of at least 0.4% to demonstrate superiority with a new antidiabetic drug in comparison to the standard therapy (but it specifies a margin of outcomes compared to the base product to demonstrate non inferiority). Such an improvement is regarded as evidence of a benefit of the given drug that makes it worth providing market approval for it. 5 Health insurance companies are also asking for “significant” improvements in glucose control when it comes to reimbursement; however, quite often they are not so sure exactly how much change of which parameter this means at the end. Health insurance companies are generally much more interested in reducing their current costs in the next few months/years even more than saving money in the long run. A reduction in the risk of developing diabetes-related late complications in the future because of an improvement in glucose control today can be a topic of lesser significance for them.
Another issue is the accuracy of dosing or measurement. Accuracy of dosing and measurement is an important safety issue. For example, insulin delivery systems such as insulin pumps and glucose meters or CGM systems must also demonstrate accuracy and precision of dose delivery and glucose measurement. 6 Where such systems are marketed, quality assurance measures are required to ensure their performance remains consistent across different batches and charges. This is important for users and also for manufacturers, who need to know what level of accuracy and precision they need to achieve for these technologies to be safe.
How to Create Evidence for Diabetes Technology?
Despite the Catch-22 situation that diabetes technology finds itself in when it comes to generating evidence, it is not advisable to avoid empirical testing of diabetes technology. Traditional methods of generating evidence include conducting RCTs for superiority and non-inferiority, or laboratory studies to assess measurement or dosage accuracy. More recently, however, new methods of data collection have become available, such as the use of RWD.
Superiority Trials
Randomized clinical trials usually aim to demonstrate the superiority of a new method or technology compared to existing treatment. The advantage of RCTs is that randomization creates observational uniformity, which helps to control most biases in trial results (Table 1). A disadvantage of RCTs is the unclear transferability of the observed efficacy of new technology into clinical care. There must be assurance that the database of patients from which the evidence was generated is similar to the general population who will be using the product.
Different Biases in Different Study Types.
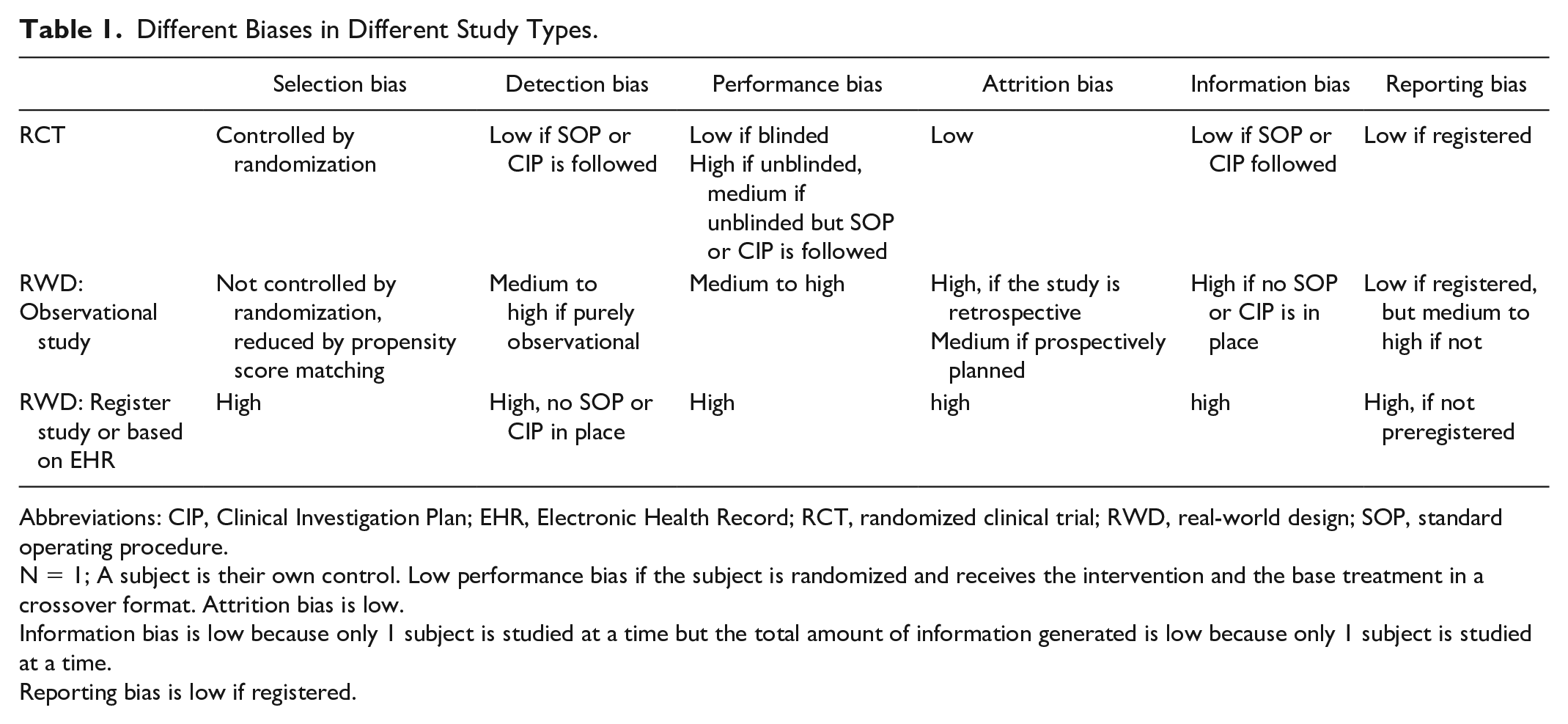
Abbreviations: CIP, Clinical Investigation Plan; EHR, Electronic Health Record; RCT, randomized clinical trial; RWD, real-world design; SOP, standard operating procedure.
N = 1; A subject is their own control. Low performance bias if the subject is randomized and receives the intervention and the base treatment in a crossover format. Attrition bias is low.
Information bias is low because only 1 subject is studied at a time but the total amount of information generated is low because only 1 subject is studied at a time.
Reporting bias is low if registered.
Non-Inferiority Trials
Non-inferiority trials may be indicated when a new method or technology can no longer be tested against the previous treatment for ethical or pragmatic reasons (eg, participants do not agree to be treated with a previous method). If such non-inferiority studies are conducted as randomized trials, then they share the same advantages and disadvantages as RCTs aiming at validating the potential effectiveness of a new technology.
Laboratory Studies
The accuracy of dosing and measurement is often investigated in laboratory studies. In such laboratory studies, for example, blood glucose measurement with a new device or system (eg, a CGM system) is compared with a reference method (eg, blood glucose measurement in the laboratory or blood glucose self-monitoring). For example, with CGM, there is currently no agreement on the reference methods used, the number of measurements and study participants required, the extent to which the accuracy of the systems is measured during provoked glucose excursions or stable glucose levels, the size and speed of such glucose excursions, and the accuracy standards in different glucose ranges. The situation is no better when it comes to dosing accuracy. 7
Real World Data
Real-world studies can also complete evidence generation. According to the FDA’s Real-World Evidence Program framework, RWD is “data related to patient health status and/or health care delivery that are routinely collected from a variety of sources.” 8 Potential sources include electronic health records, medical claims and billing data, data from product and disease registries, patient-generated data, including from the home, and data from other sources that can provide information about health status, such as mobile devices. The advantage of RWD is clearly that they are obtained in routine care so that the transferability of study results to clinical reality does not arise here.
These RWD can be used to generate Real World Evidence (RWE), which is “clinical evidence about the use and potential benefits or risks of a medical device derived from analysis of RWD.” Although the FDA is generally open to the use of RWD and the generation of RWE for specific purposes, the agency is aware of the possibility of bias in RWD-based effect estimates. In particular, the agency is aware of potential confounding bias in observational studies, registry data, or electric patient records,8,9 A confounding factor is a pre-intervention prognostic factor that predicts whether an individual receives one or the other intervention of interest. Typical confounding bias can be the severity of pre-existing disease, presence of comorbidities, health care utilization, or socioeconomic status. Often more motivated or better-educated individuals choose to be involved in creating RWD by participating in registries or uploading sensor or treatment data to the appropriate database, which may lead to an overestimation of an intervention effect. The assessments recorded in electronic health records, health insurance claims data, and registries may also vary among different health professionals because they use different outlines or no Subjective-Objective-Assessment-Plan (SOAP) outline at all for assessments or documentation. In general, RWE data compared to RCT data produce a greater quantity of data but each individual piece of data may be of lower quality. Real world evidence trials are useful as confirmatory evidence of a product’s benefit and are also useful for identifying rare side effects.
N of 1 Trials
N-of-1 trials are a type of real world data consisting of a set of multiple crossover trials, that are randomized and sometimes blinded, conducted in a single subject. 10 In the past few years, since databases from the electronic health record have become widely available, there has been less interest in performing trials on 1 patient at a time and more interest in assessing large populations form databases.
What Evidence Is Needed for What?
In Table 2, some thoughts are listed about which form of evidence is generated by which study design and type. Qualitative, disruptive, and new diabetes technologies, such as the CGM systems or AID systems, should be (and are) tested for superiority in comparison to existing treatment options employing RCT. Because of the randomization, this type of study provides the best guarantee for the control of bias in the study results. In the case of a significant and clinically relevant effect, it is of course easier to convince the payers of a higher reimbursement than for the previous therapy.
Which Form of Evidence is Generated by Study Design and Study Type?
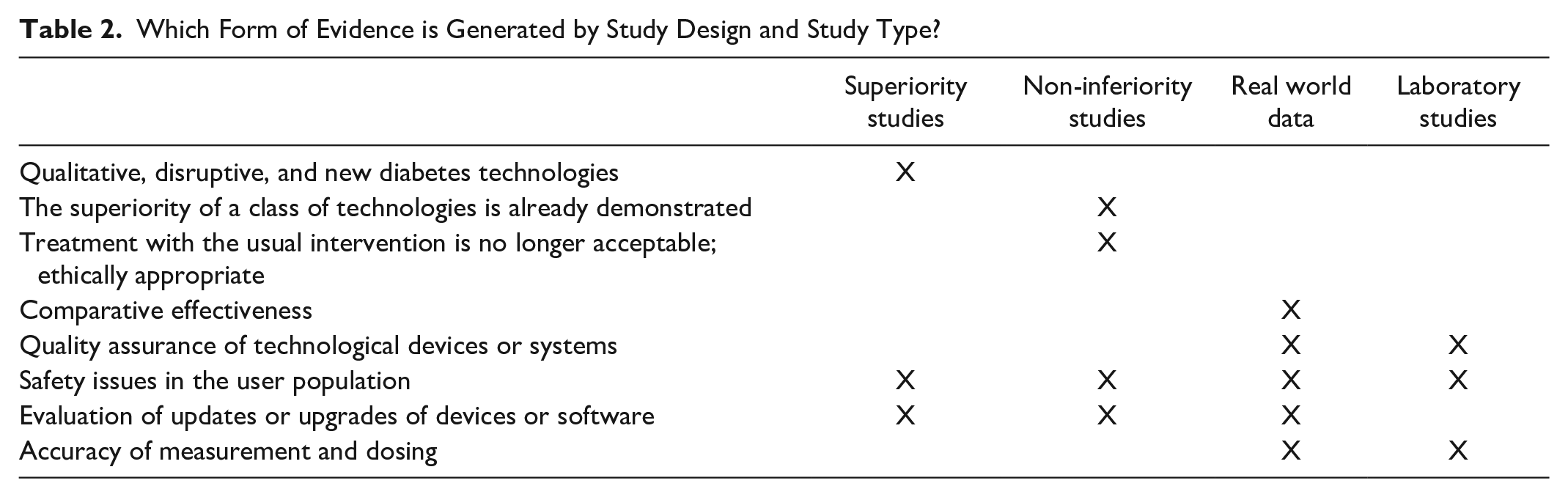
Once a particular diabetes technology has been established, it may be difficult or even unethical to find subjects for the control group who are still using the former standard treatment. One example is new glucose monitoring technologies that can replace traditional blood glucose self-monitoring. Especially in PwD with type 1 diabetes, it is difficult to recruit a control group still using Self Monitoring of Blood Glucose (SMBG) for studies with a new CGM approach. Newer developments within the same class of diabetes technology could therefore be tested with non-inferiority studies in comparison to an established system or device. If other advantages come into play, such as lower price, higher reliability, or fewer adverse events, then an advantage for clinical care can also be inferred.
Real-world design and studies based on these data are primarily suited to ensure that new technology established in clinical care has a comparative efficacy as tested with RCT or non-inferiority studies in routine clinical care. Furthermore, RWE can also ensure that quality standards are persistently met concerning clinical efficacy. In addition, rather rare safety issues can be more reliably detected in a larger population with RWE than in randomized controlled trials or non-inferiority trials, which are powered to detect superiority or non-inferiority concerning a predefined effect size in a thoroughly analyzed relatively small population. Upgrades of technological systems or devices or updates of software usually do not necessarily require testing for superiority or non-inferiority, especially if they involve purely technical improvements: Here, too, real-world studies could provide timely evaluation results.
Of course, the types of evidence generation studies are not as easily distinguishable as the table might suggest. For example, it is not always clear when an upgrade of a device or system or an update of software implies a better quality treatment that should be evaluated by an RCT or a noninferiority trial rather than a real-world study.
Conclusion
The evaluation of evidence of a given diabetes technology is subject to a so-called Catch-22 dilemma, which is characterized by the fact that, on the one hand, a methodologically sound evaluation is important, but on the other hand, the speed of technological progress does not always allow an evaluation with the help of RCTs.
The purpose of this editorial is to stimulate a discussion among scientists, regulators, manufacturers, and people with diabetes about which standards of evidence are appropriate for which innovations in diabetes technology. Table 2 should be seen as a starting point for such a discussion, not an end point. We believe that such an agreement can accelerate market access for new diabetes technologies by avoiding unnecessary evidence requirements for the evaluation of updated technologies, thereby reducing costs. At the same time, such an agreement can also help to provide important evidence of the efficacy, safety and usability of new diabetes technologies for people with diabetes, health care professionals, scientists, regulatory bodies, and health insurance companies.
Footnotes
Acknowledgements
Helpful comments by Dr. Guido Freckmann about accuracy of measurement were very appreciated.
Abbreviations
CGM, continuous glucose monitoring; RCT, randomized clinical trials; RWD, real world data; RWE, real world evidence; SOAP, subjective-objective-assessment-plan
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: NH is member of the advisory board for Insulet, Research support from Roche Diagnostics, Sanofi, Embecta
BK is a member of the advisory board of Roche Diabetes Care, Sanofi-Aventis, NovoNordisk, Berlin Chemie, BD and Insulet. He received speaker honoraria from Roche Diabetes Care, Sanofi-Aventis, NovoNordisk, Berlin Chemie, Decxom, BD, Abbott Diabetes Care and Insulet.
DCK is a consultant for EOFlow, Integrity, Lifecare, and Thirdwayv.
LH is a consultant for several companies that are developing novel diagnostic and therapeutic options for diabetes treatment. He is a shareholder of the Profil Institut für Stoffwechselforschung GmbH, Neuss, Germany.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.