
Abstract
Modern changes in diet and lifestyle have led to an explosion of insulin resistance and metabolic diseases around the globe which, if left unchecked, will become a principal driver of morbidity and mortality in the 21st century. The nature of the metabolic homeostatic shift within the body has therefore become a topic of considerable interest. While the gut has long been recognized as an acute nutrient sensor with signaling mechanisms to the other metabolic organs of the body, its role in regulating the body’s metabolic status over longer periods of time has been underappreciated. Recent insights from bariatric surgery and intestinal nutrient stimulation experiments provide a window into the adaptive role of the intestinal mucosa in a foregut/hindgut metabolic balance model that helps to define metabolic parameters within the body—informing the metabolic regulation of insulin resistance versus sensitivity, hunger versus satiety, energy utilization versus energy storage, and protection from hypoglycemia versus protection from hyperglycemia. This intestinal metabolic balance model provides an intellectual framework with which to understand the distinct roles of proximal and distal intestinal segments in metabolic regulation. The model may also aid in the development of novel disease-modifying therapies that can correct the dysregulated metabolic signals from the intestine and stem the tide of metabolic diseases in society.
Introduction
The Type 2 Diabetes (T2D) epidemic is global and growing. Despite broad availability and access to lifestyle therapies and multiple classes of anti-diabetic medications, the vast majority of patients with T2D still do not achieve optimized glycemic control or risk factor reduction. In fact, fewer than one third of patients with T2D achieve and maintain the generally accepted HbA1c goal of 7% or less for adult T2D patients. 1 A marked discrepancy has emerged between increasingly impressive results from controlled clinical trials of new drugs and stagnant real-world outcomes in disease treatment. This difference is principally due to fundamental gaps in real world outcomes that current drug therapies have not been able to fill: excessive dependence on adherence, 2 lack of durable disease modification, 3 therapeutic inertia from polypharmacy fatigue, 4 and the cost of novel pharmacotherapies.
It is evident that one important root cause of metabolic disease is the impact of modern diets on our body’s metabolic control systems. Recent observations of durable T2D remission from bariatric surgery provide a better appreciation for the critical role of the gut in nutrient sensing and metabolic control.5,6 We present a theory of a causal mechanism linking nutrient-induced alterations of the gut mucosa to gut neuro-hormonal signaling changes and the metabolic syndrome, which we call the Intestinal Metabolic Balance model. We believe this model provides a useful intellectual framework with which to understand the role of intestinal mucosal signaling as an important mediator of the metabolic dysregulation caused by modern diets. The model also provides a mechanistic explanation for some aspects of the metabolic benefits of certain bariatric surgeries and points to novel disease modifying therapies targeting the gut which have the potential to reduce the metabolic disease burden in society. The model will hopefully prove to be useful in helping to shed light on critical areas where more research is surely needed to better explicate the role of the gut in metabolic dysfunction.
A New Paradigm Emerges from an Unlikely Origin
Metabolic surgeries, originally intended for weight loss, have emerged as a treatment approach in T2D with superior metabolic benefits compared to the current standard of care.7,8 Despite the profound metabolic benefits observed after surgery, little is known about the multiple mechanistic underpinnings leading to these benefits. Interestingly, physiological studies in people undergoing bariatric surgery have surprisingly revealed that many of the metabolic improvements occur before substantial weight loss and these early improvements appear to be independent of the degree of weight loss achieved. 9 In particular, the exclusion of the duodenum from the passage of nutrients by surgical diversion results in weight-independent metabolic improvements associated with a reduction in insulin resistance.10-12 This effect is notable in its almost immediate onset (within days) and in its reversibility (such as by re-exposure of the duodenum to nutrients). In 2010, Holst and colleagues examined the glucose tolerance of a patient with T2D who underwent a Roux-en-Y gastric bypass surgery (RYGB) and who happened to need a percutaneous G-tube placement directly into the bypassed stomach to enable feeding via the duodenum after the surgery. 13 In this patient, oral delivery of glucose (bypassing the duodenum) led to a markedly superior metabolic profile compared to G-tube delivery of glucose (enabling nutrient transit through the duodenum as if the patient had not had surgery) despite both procedures being performed in this individual at the same weight and on successive days (Figure 1). Bypass of the proximal small bowel, and the resulting increased nutrient delivery to the distal intestine, thus appeared to improve glucose homeostasis independently from calorie malabsorption or weight change.14,15
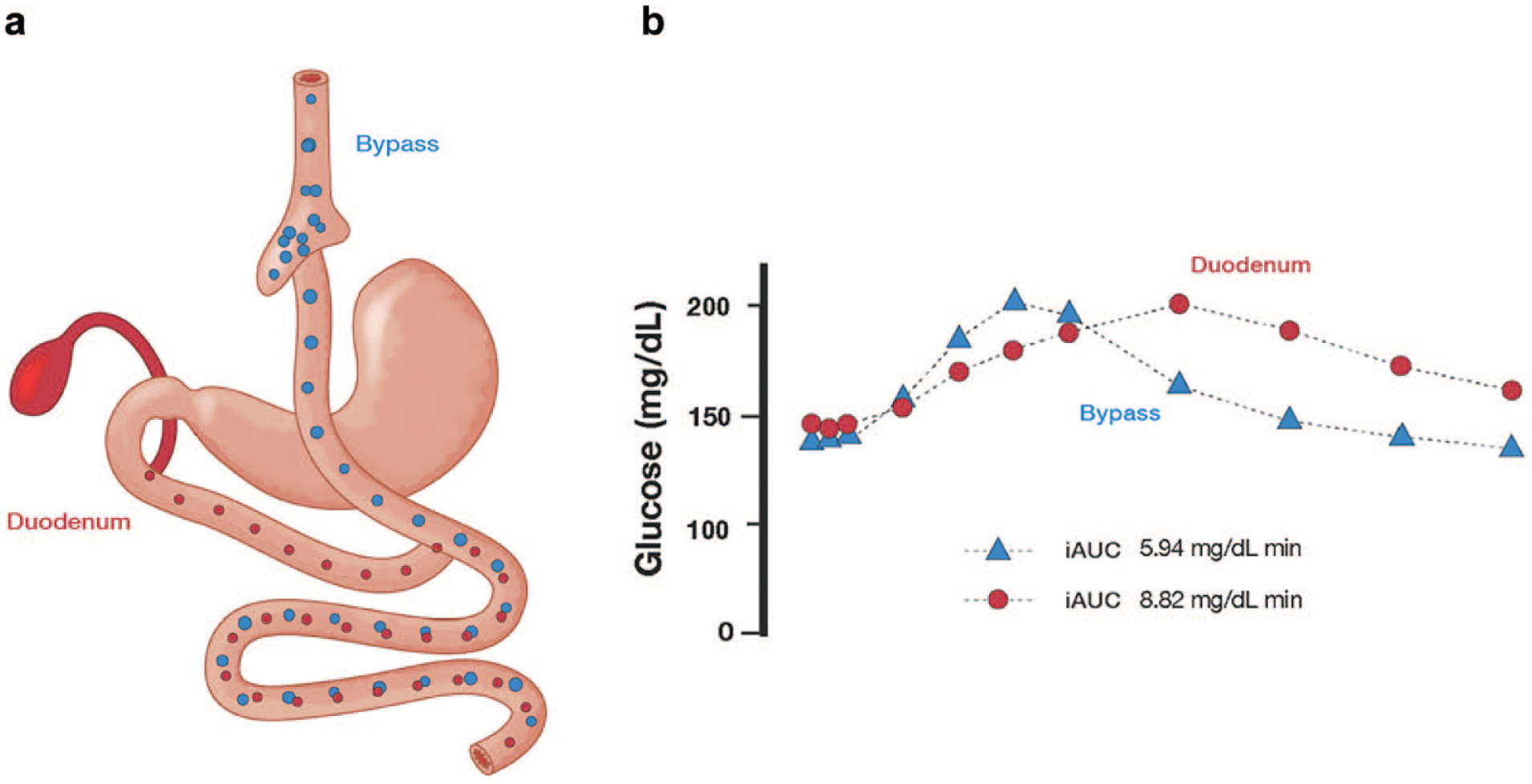
Glucose Metabolism Studies after RYGB. (a) Diagrammatic representation of RYGB surgery, in which a small gastric pouch is connected surgically to the mid-jejunum, leaving the majority of the stomach, the entire duodenum, and some of the proximal small intestine excluded from the passage of nutrients. Bile acids and pancreatic enzymes continue to pass through the bypassed segment of the small intestine (red dots) while ingested food passes from esophagus through a small gastric pouch and into the connected jejunal portion of the small intestine (blue dots). Reprinted with permission from Blamb/Shutterstock.com. (b) Oral glucose tolerance test with nutrient delivery via the bypassed stomach and duodenum (red circles) versus by the gastric pouch and connected jejunal portion of the small intestine (blue triangles) shows markedly different glucose tolerance curves and higher glucose excursion with duodenal nutrient delivery compared to delivery via the bypass. Reprinted with permission from Dirksen et al. 13
These observations have led many to ask if this immediate metabolic benefit is a neurohormonal consequence of preventing duodenal nutrient exposure (the “foregut” theory) or enhancing distal small intestinal nutrient exposure (the “hindgut” theory). 9 There are several important pieces of evidence arguing for the metabolic benefits of preventing nutrient contact with the duodenum. In one set of experiments with animal models, it has been shown that surgically constructing a gastro-jejunal anastomosis while still maintaining a nutrient pathway to the duodenum is not as metabolically favorable as a gastro-jejunal anastomosis in which the duodenum is totally excluded. 11 Additionally, some humans who undergo a RYGB surgery in which the distal stomach and duodenum are excluded via a surgical closure of the stomach present later with the incidental surgical complication of a gastro-gastric fistula which once again allows nutrients to be delivered to the proximal intestine. These patients often present with deteriorating glucose control after RYGB, which can be corrected by closing the gastro-gastric fistula and excluding the duodenum from nutrient delivery once again.16,17 Collectively, these surgical studies all point to a potent, weight-independent effect on systemic glucose control associated with altering the location of nutrient delivery within the intestine in a manner that excludes foregut nutrient exposure and consequently increases hindgut nutrient exposure.
Many groups have since tested the metabolic effects of altering the location of nutrient contact in the intestine via non-surgical approaches. Investigators have shown in humans with T2D that the luminal delivery of glucose to the proximal intestine results in worse control of glucose metabolism than the same nutrients delivered to the distal small intestine.18,19 Duodenal delivery is associated with worse insulin resistance and higher rates of lipolysis than nutrient delivery to the mid-jejunum. 20 Others have shown that oral ingestion of slowly digestible sugar isomers (which are absorbed predominantly in the distal small intestine rather than the duodenum) lead to better glucose homeostasis than equi-caloric sucrose or fructose (which are absorbed more proximally). 21 Taken together with the surgical studies, these disparate experiments defy much of the conventional wisdom in T2D and nutritional science by showing that the location of nutrient exposure in the intestine, in addition to known contributions of genetics, activity level, caloric content, and nutritional status can also have a profound impact on glucose homeostasis.
Moreover, surgical bypass has demonstrated quite convincingly that intestinal re-routing not only impacts metabolism acutely during a meal, but also alters baseline insulin resistance and metabolic disease. In a rodent diabetes model, duodenal bypass improves fasting glucose and glucose tolerance. 10 In diet-induced obesity (DIO) models, duodenal bypass improves insulin sensitivity and reduces hepatic fat accumulation compared to sham-operated, weight-matched animals, independently of weight loss or reduced caloric intake. 22 A more comprehensive model is needed to account for the deleterious effects of duodenal nutrient delivery and the beneficial effects of distal intestinal nutrient delivery on basal levels of insulin sensitivity and glucose homeostasis in obesity and T2D.
The Gut as the Body’s Metabolic “Balance”
In addition to its better appreciated role in nutrient absorption, the gastrointestinal tract is the largest endocrine organ in the body and contains an important microbiome and an enteric nervous system. The enteric nervous system includes neural circuits that modulate motor functions, local blood flow, mucosal transport and secretion, and immune and endocrine functions. The lining of the intestine serves as both a food-sensing and hormone-secreting interface for the body. Its mucosal lining is comprised of several cell types, approximately 1% of which are enteroendocrine cells (EECs) that release metabolically active hormones regulated by specific nutrient absorption (or lack thereof) in a finely tuned pattern to help the body maintain adequate fuel supply and metabolic homeostasis in both the fasting and post-prandial states. EECs are also neuroendocrine cells, synaptically communicating with the enteric nervous system via neuropods at their basement membrane and thereby transmitting signals related to nutrient status.23,24 Critically, nutrient absorption and nutrient-induced neurohormonal signaling are inextricably linked to one another by the transport of nutrients across the luminal brush border.6,25 Because of this, nutrient “sensing,” nutrient absorption, and nutrient-induced neurohormonal impact can be viewed interchangeably. Different EECs along the length of the GI tract secrete different hormones, thus the sensory impact of fuel is regulated not only by the type of nutrients sensed by the mucosa but also the location in the GI tract where those nutrients are sensed. 21
Why is it that the location of nutrient delivery in the intestine can have such a profound impact on metabolic control? One possibility is that the fundamental abnormality of intestinal metabolic control is one that occurs at the organ level rather than the cellular or molecular level. The intestine in adult mammals is an organ that is known to be able to homeostatically grow or shrink over time to adaptively resize in response to varying environmental conditions (eg, changes in nutrient delivery). 26 We hypothesize that diets that are high in easily digested fats and sugars represent an important driver of the adaptive resizing of the human intestine, causing mucosal alterations of the small intestine through the stimulus of heightened nutrient delivery. This adaptation leads to hyperplasia of the proximal intestinal mucosa and the simultaneous hypoplasia of the distal intestinal mucosa. 27 In this view, the mucosal adaptation of the proximal intestine to high fat and sugar diets leads to a morphological alteration that correlates with the alterations in neurohormonal signal from the proximal intestine, accelerated nutrient absorption via the proximal intestine, and a reduced basal neurohormonal signal from the distal intestine. 19 The relationship between structural changes of the intestinal mucosa and dysfunctional changes in neurohormonal signaling from the proximal intestine to the rest of the body remains an open question. We believe the net systemic impact of this diet-induced imbalance of proximal/distal intestinal neurohormonal signals in both the basal and post-prandial states may well be the basis for the dysregulated neurohormonal impact of the gut on the metabolic status in obese and insulin resistant individuals.
Intestinal surgeries that shift the balance of nutrient exposure from the proximal to the distal intestine (including certain bariatric surgeries) achieve weight-independent improvements in metabolic disease control. 9 Moreover, the heightened distal nutrient delivery that invariably occurs as a consequence of duodenal bypass leads to an expansion of the distal intestinal mucosa through a similar process of mucosal adaptation, further amplifying the shift toward more distal hormonal factors after surgical bypass. 28
Mechanistic studies of duodenal bypass surgery or catheter-based nutrient diversion have highlighted a reduction of adipocyte lipolysis as a potential mediator of this metabolic improvement, perhaps driven by gut-brain signals, gut hormone effects, bile acid signaling, microbiome effects, or combinations of these.20,29-31 This integrated signal is likely achieved via a myriad of effector pathways. It may therefore be better understood by the net effect of cumulative proximal versus distal signals than by the individual effects of any of the signaling molecules therein.
It is remarkable that, despite the amount of evidence supporting the role of the intestine in metabolic control, the precise mechanisms of this control remain elusive. In Figure 2 we show our proposed model of the gut lining as a longitudinal “metabolic balance” for the body and mucosal adaptation as a cellular mechanism within the mucosal lining by which this balance can be altered. Mature individuals who consume a healthy diet have a normal metabolic status with balanced contributions from proximal and distal intestinal neurohormonal signals (Figure 2a) to the body’s basal and post-prandial metabolic state. Individuals who consume diets that are high in fat and sugar (and presumably low in fiber) are predisposed to developing dysfunction of the proximal intestinal mucosa because of the proximal intestine’s adaptation to chronically elevated nutrient exposure. This shifts the metabolic “balance” toward the proximal intestine (with subsequently reduced distal nutrient delivery) and pushes the metabolic state toward hunger, energy utilization, and insulin resistance, all in the setting of excess caloric availability (Figure 2b). Conversely, individuals who undergo duodenal bypass surgery increase distal nutrient delivery, which leads to hypertrophy and hyperplasia of the distal intestinal mucosa. This, in turn, shifts the metabolic balance toward the distal intestine and pushes an individual’s homeostatic status toward satiety, energy storage, and insulin sensitivity and also contributes to weight loss and improved metabolic control. (Figure 2c).
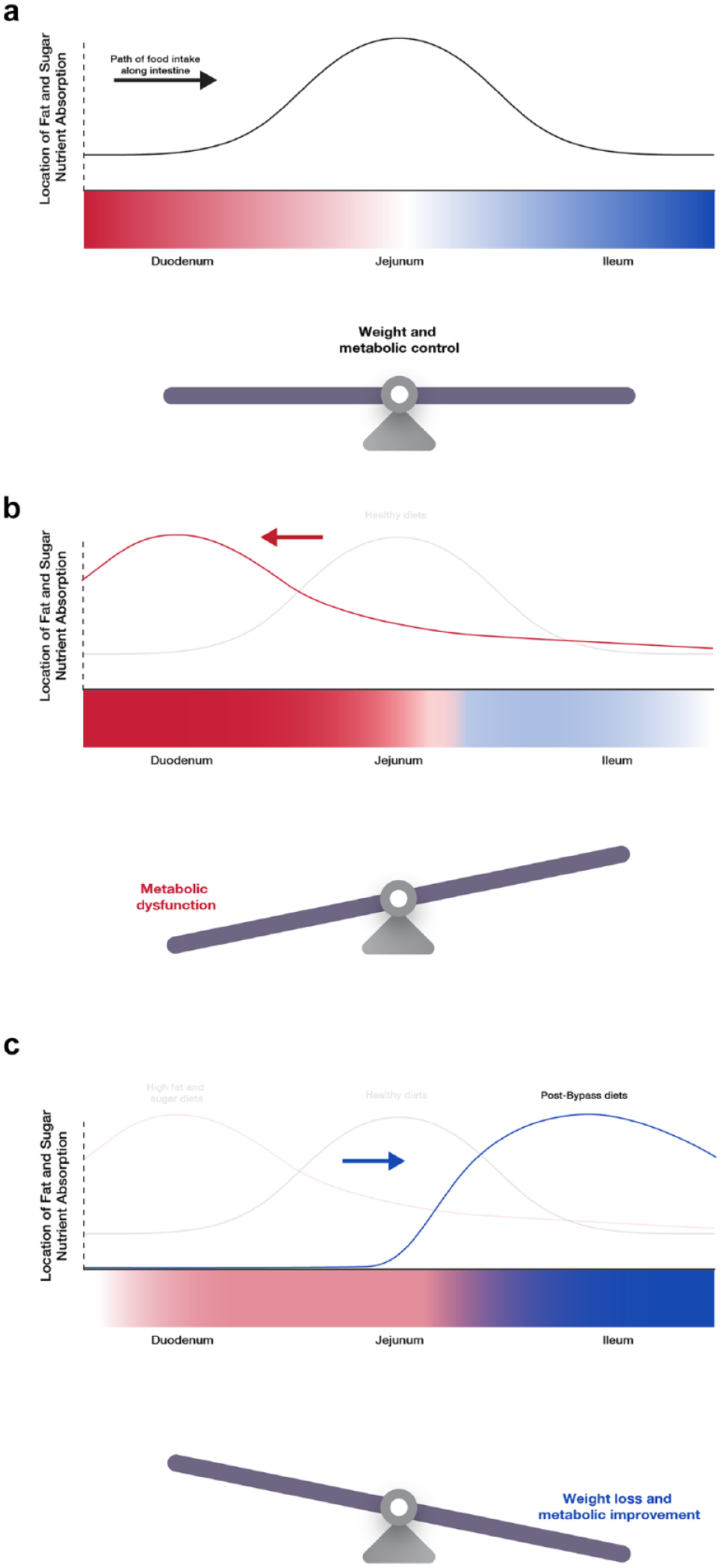
Metabolic Balance Model of Glucose Homeostasis. (a) The normal path of food intake along the length of the GI tract is from the proximal intestine to the distal (ie, from the duodenum to the jejunum and then the ileum). Nutrient absorption of healthy diets that are relatively scarce in simple sugars and fats occurs as food is digested along the length of the GI tract, with relatively limited micronutrient absorption in the duodenum and a larger amount of absorption in the mid and distal small intestine. This allows for nutrient sensing and hormonal release from the proximal, mid, and distal intestine, helping enable the body to maintain homeostasis of hunger vs satiety and energy utilization (fat breakdown) versus energy storage. (b) Diets that are high in simple fats and sugars are more easily digested and absorbed in the proximal intestine, causing duodenal hyperplasia and increased proximal neurohormonal signaling. This shifts the gut’s metabolic balance toward hunger and fat breakdown, contributing to increased obesity and insulin resistance. (c) RYGB and similar bariatric surgeries prevent or limit nutrient absorption in the proximal intestine and accelerate delivery of nutrients to the distal intestine, leading to increased distal intestinal neurohormonal signaling. This shifts the gut’s metabolic balance toward satiety and energy storage, contributing to weight loss and improved metabolic control.
Evidence for the Metabolic Balance Model
Experimental data from rats and humans demonstrate that prolonged exposure to a Western diet can lead to an expansion of the duodenal mucosa and an increase in enteroendocrine cell numbers in this region.32,33 We and others have observed in animal models a hyperplasia of the duodenal lining in response to diets high in fats and sugars. 27 This duodenal hyperplasia is associated with deeper crypts, greater crypt density, longer mucosal villi, more duodenal mucosal cells of all differentiated types, and greater mucosal surface area. Experiments with human duodenum-derived intestinal organoid cultures reveal that the direct exposure of duodenal mucosal stem cells to higher concentrations of fat and sugar leads to higher rates of stem cell division, thereby connecting nutrient exposure to the known acceleration of stem-cell division underlying intestinal mucosal adaptation. This adaptation to nutrient exposure may explain how diets that are high in easily digestible fats and sugars, as seen in modern society, may yield a hyperplastic proximal intestinal mucosa as a direct consequence of heightened local nutrient exposure. 26 It is important to note that although organoid cultures are suitable for cellular mechanistic and developmental studies, data generated from this model system should not be overinterpreted as representative of organism-level complex physiological mechanisms. Further studies are needed to better understand the nature and time course of duodenal adaptation and dysfunction.
Duodenal hyperplasia appears to be linked to insulin resistance and the metabolic syndrome, which is a precursor to Type 2 Diabetes, non-alcoholic fatty liver disease, and obesity. While the precise molecular consequences of duodenal dysfunction are not known, increased Gastic Inhibitory Polypeptide (GIP) secretion, increased iron absorption, altered gut-brain signals, bile acid changes, and microbiome alterations have all been invoked as potential mechanisms.23,34,35 GIP is an incretin hormone that contributes to the basal secretion of insulin and glucagon in the fasting state as well as their induced secretion post prandially. 34 It is produced in intestinal cells found predominantly in the proximal small intestine, including the duodenum. In addition to its effects on the alpha cell and beta cell of the pancreas, GIP also has stimulatory effects on both adipose tissue lipogenesis and lipolysis through both direct and indirect means.32,36 In animal models fed a high fat diet, GIP expression was increased, as was intestinal K cell number. 34 Observations of GIP excess extend to humans as well. Elevated levels of GIP have been seen in serum from obese patients 34 and in biopsy samples of duodenal mucosa taken from patients with T2D. 37 It is evident that GIP expression is increased in people with obesity and T2D, but the role of pharmacologic manipulation of the GIP signaling pathway in metabolic control is unclear, with both GIP agonists and antagonists currently in development.38-40 In addition, the literature linking GIP to the development of metabolic disease must be interpreted with caution, given that studies have also proposed that GIP may have an insulin-sensitizing effect. 41 Iron absorption occurs primarily in the proximal small intestine, including the duodenum. Also similar to GIP, elevated iron stores are positively associated with insulin resistance, metabolic syndrome, and T2D in humans. 42 The relationship between iron and diabetes is reciprocal: elevated iron stores negatively impact glucose metabolism and glucose metabolism impacts iron pathways in the body. Iron excess predicts the development of T2D while iron depletion appears to protect from metabolic disease. The intestine has also been observed to have alterations in gut-brain, bile acid, and microbiome signaling in animal models and individuals with obesity, as has been reviewed elsewhere.6,23
It is also possible that duodenal hyperplasia does not confer any afferent signal itself but simply acts as a hyperactive accelerant of nutrient uptake and a “gatekeeper” that limits nutrient delivery to the distal gut. When the duodenum of rodents fed a high fat and sugar diet becomes hyperplastic, the distal intestine of the same DIO rodents becomes hypoplastic, which is hypothesized to be due to reduced delivery of nutrients to the distal small intestine. Physiologic characterization of humans and rodent models of T2D and obesity has been associated with decreased GLP-1 and Peptide YY (PYY) secretion that correlate with distal hypoplasia.43-45 As with the link between proximal intestinal hyperplasia and GIP excess, strong circumstantial evidence also supports an important hormonal consequence of distal intestinal hypoplasia. As such, the “foregut” mechanism may simply be a mechanism by which duodenal hyperplasia reciprocally diminishes known hindgut effects on satiety and glucose reduction mediated through distal effectors, including GLP-1 and PYY. In summary, while the evidence that duodenal hyperplasia may be pathologically implicated in insulin resistance is quite strong, the afferent mechanistic relationship between duodenal hyperplasia and the metabolic syndrome remains unclear.
The growing body of evidence around gut-segment dependent metabolic effects is not sufficiently explained by existing models of the role of the gut in metabolic control. Our model adds to, but does not replace, our growing understanding of the key integrating roles played by adipose tissue, the brain, and other organs as critical participants in determining metabolic homeostasis. 46 For instance, current theories recognize the role of incretin hormones and the microbiome on insulin kinetics and the role of nutrient signals from the gut in influencing the brain-adipose and brain-liver signaling required for energy mobilization versus energy storage. 47 However, they do not explain the contrasting effects of proximal intestinal and distal intestinal factors on weight, lipolysis, insulin resistance, glycemia, or cardiovascular risk factors. 48 It is possible that the intestinal metabolic imbalance caused by a hyperplastic duodenum drives accelerated fatty acid flux (either through neuronal or hormonal afferent signals), thereby tying the mucosal abnormality to the development of insulin resistance. We believe this gut mucosal metabolic balance model provides a rational explanation for the histological observation of mucosal changes in response to high fat diets along the length of the GI tract, as well as such clinical observations as: (1) the weight-independent benefits of certain duodenum-excluding surgeries, (2) the different responses of nutrient stimulus in the proximal versus distal intestine, (3) the benefits of GLP-1 receptor analogues, and even (4) the benefit of alpha-glucosidase inhibitors (because inhibition of proximal intestinal glucose absorption leads to enhanced distal glucose absorption). Furthermore, the model provides a framework to help interpret the mechanistic basis of the human clinical evidence thus far available and enables the generation of hypotheses that can provide a path forward to new treatments.
There are several unique novel endoscopic interventions under development that have each demonstrated weight independent metabolic benefits. While working via disparate mechanisms, they each harness the potential to reset this putative metabolic imbalance in a minimally invasive and adherence-independent manner by altering the location of intestinal nutrient absorption. Three categories of such promising interventions are worth considering. First, the duodenal-jejunal bypass liner, which is a luminal implant that causes nutrient delivery to bypass the duodenum and proximal jejunum, thereby accelerating nutrient delivery to the distal intestine, has shown promising effects on blood glucose control and weight loss.49,50 Second, the jejuno-ileal partial bypass procedure accelerates nutrient delivery to the distal intestine, 51 which has been shown to reduce blood sugar and body weight in individuals with T2D. 52 Third, duodenal mucosal resurfacing (DMR) addresses dysfunctional duodenal neurohormonal signaling via hydrothermal ablation, leading to improved insulin sensitivity and glucose control. 49 Early studies of each of these therapies have also shown decreases in insulin resistance, liver fat content, and body weight, thereby supporting the argument that intestinal interventions show considerable promise in reversing metabolic diseases.
Conclusion
While there are multiple dynamic physiological mechanisms that contribute to the maintenance of metabolic homeostasis, this metabolic balance model of the gut should be viewed as a starting point for further hypothesis generation and testing. What is the link between duodenal dysfunction and insulin resistance? What is the nature of the afferent signal from the gut to the brain, liver, and/or adipose that regulates the balance between lipogenesis and lipolysis and impacts the systemic insulin resistant state? Is there a minimal reducible set of proximal intestinal and/or distal intestinal factors that are sufficient to recapitulate the phenotype of bariatric surgery in a molecular manner (i.e., what are the contributions of gut hormones, bile acids, iron metabolism, neuronal signaling, and the microbiome involved)? We have recently convened a research consortium of scientific and clinical experts to begin delving into these potential questions as a means to better understand the role the gut is playing in metabolic homeostasis.
Though the precise molecular mechanisms may still be unknown, it is possible that entirely new therapeutic approaches can be developed based on this gut metabolic balance model. These approaches may well address the current explosive unmet need for disease modifying therapies by providing non-pharmacological, adherence-independent alternatives to the current standard of care in T2D. Minimally invasive, highly scalable approaches aiming to restore intestinal metabolic equipoise have the potential for far-reaching consequences on human health by tackling one of the upstream, organ-level root causes of the metabolic syndrome rather than simply addressing its narrower downstream consequences, like elevated blood sugar, liver steatosis, or coronary atherosclerosis.
Footnotes
Acknowledgements
The authors wish to thank Emily Cozzi, PhD and Kelly White, PharmD for their valuable input and assistance in the writing and technical editing of this manuscript.
Abbreviations
Diet-induced obesity (DIO), Enteroendocrine cells (EECs), gastrointestinal (GI), Gastric Inhibitory Polypeptide (GIP), Peptide YY (PYY), Roux-en-Y gastric bypass (RYGB), (T2D) Type 2 Diabetes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Harith Rajagopalan and Juan Carlos Lopez-Talavera are full time employees of Fractyl Health, Inc. David Klonoff is a consultant for EoFlow, Fractyl Health, Inc., Lifecare, Novo, Roche Diagnostics, Samsung, and Thirdwayv. Alan Cherrington is a consultant to Fractyl Health, Inc., Abvance, Biocon, Diakard, Metavention, Novo Nordisk, Sekkei Bio, Sensulin Labs, Thetis Pharmaceuticals, vTv Therapeutics. Dr. Cherrington has research contracts with Abvance and Novo Nordisk.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Fractyl Health, Inc.