
Abstract
Importance
Nasopharyngeal carcinoma (NPC) is closely linked to microorganisms, especially intra-tumoral microbiota. However, the role of commensal microbiota in NPC remains underexplored, with implications for understanding disease mechanisms.
Objective
This study aims to analyze and compare the bacterial microbiota in the nasopharynx and middle meatus (MM) of individuals with NPC and those without NPC. Additionally, the study seeks to identify potential microbial biomarkers that can distinguish between NPC and non-NPC (nNPC) individuals.
Design
Cross-sectional study.
Setting
Study conducted in a clinical setting with NPC and non-NPC participants to evaluate microbial diversity relevant to NPC.
Participants
Ten NPC cases and 15 non-NPC controls were recruited based on clinical eligibility.
Main Outcome Measures
Bacterial microbiota sampling from the nasopharynx and MM was analyzed by 16S rRNA sequencing. Microbiota diversity (alpha and beta diversity indices), presence of bacterial taxa with biomarker potential, and prediction model accuracy [area under the curve (AUC)].
Results
Microbiota diversity was significantly lower in NPC patients versus controls. In nasopharyngeal samples, alpha diversity (Chao1 index, P = .02) and beta diversity (PERMANOVA, P = .001) differed notably between groups, though MM samples showed no significant difference (Chao1 index, P = .23). Machine learning identified Pseudomonas, Cutibacterium, and Finegoldia as potential NPC biomarkers (AUC = 0.86).
Conclusions and Relevance
This pioneering study highlights dysbiosis in nasopharyngeal microbiota among NPC patients. Findings suggest that Pseudomonas, Cutibacterium, and Finegoldia may be useful biomarkers for NPC diagnosis, warranting further investigation into microbial roles in NPC pathogenesis.

Background
Microbiota is closely associated with cancer development, progression, and treatment outcomes through mechanisms such as immune regulation, metabolic pathways, and microbial diversity, although the exact mechanisms remain largely unknown1,2-4 It is universally recognized that around 17% of cancers in humans worldwide result from microbes. 3 Saliva microbiota is considered an important biomarker for head and neck squamous cell carcinoma. 5 Airway microbiome plays a role in lung cancer initiation and progression, potentially triggering tumor initiation and progression through the production of bacterial toxins and proinflammatory factors. 6 Respiratory microbiota composition significantly differs between lung cancer patients and healthy individuals and may be used as a potential biomarker of lung cancer. 7 Nasopharyngeal carcinoma (NPC), is an uncommon respiratory cancer, and the link between it and Epstein-Barr virus (EBV) has been firmly established. 8 However, the relation among commensal microbiota, EBV, and NPC still needs more evidence.
NPC is a relatively uncommon form of upper airway cancer worldwide, with an incidence of less than 1 per 100,000 person-years which comprises primarily external-beam radiation therapy with or without concurrent cisplatin-based chemotherapy.9,10 Endoscopic nasopharyngectomy is considered a treatment modality for early-stage recurrent nasopharyngeal carcinoma (rNPC). 11 Nevertheless, incidence rates in Taiwan range from 2.8 to 6.6 per 100,000 person-years, which is significantly higher than in other countries.9,12 As previously noted, the microbiota is strongly linked to cancer development, but additional evidence is needed to determine the exact connection between NPC and microbiota. Zhong et al presented compelling data indicating a correlation between the tumor microbiome and NPC. 4 Their findings suggest that the tumor microbiome has the potential as a biomarker for early NPC diagnosis and prognostic indicator, including Turicibacter. 4 Qiao et al have identified intra-tumoral bacterial load as a reliable prognostic indicator for patients with NPC, indicating the potential use of this marker as a guide for treatment decisions in patients at varying degrees of malignant progression risk. 1 Both studies provide significant evidence of the relationship between intra-tumoral microbiota and NPC. However, the commensal microbiota of NPC and its bacteriological characteristics in the nasopharynx, as well as their impact in other nasal locations, remain unknown. Therefore, in this study, a culture-independent approach involving 16S rRNA gene amplicon sequencing was used to compare the microbiota composition between NPC and non-NPC cases and to determine the relationship between the involved bacteria.
Methods
Study Design and Population
A cross-sectional study was undertaken at Chung Shan Medical University Hospital, involving patients newly diagnosed with NPC and a control group without NPC [non-NPC (nNPC)]. Eligible participants were all aged above 20 years and had provided informed consent before the collection of samples. Inclusion criteria stipulated that the patients within the NPC group underwent endoscopic nasopharyngeal tumor biopsies conducted by otolaryngologists. The diagnosis of NPC was confirmed according to the World Health Organization classification 13 by 2 pathologists. Conversely, participants in the control group (nNPC) underwent endoscopic evaluations by otolaryngologists, revealing no nasopharyngeal tumor formation.
Participants in both the NPC group and the control group without NPC were excluded based on the following criteria: a history of head and neck cancer, previous treatment with radiotherapy or chemotherapy, acute rhinosinusitis 14 or chronic rhinosinusitis 14 with acute exacerbation within the last month, cystic fibrosis, immunocompromised status, administration of systemic or topical antibiotics or antifungal medications within 1 month preceding the study, functional endoscopic sinus surgery, septomeatoplasty, skull base surgery, nasopharyngectomy, adenoidectomy within 1 year, the presence of severe oral diseases (such as oral fungal infections and oral cancer) within 1 month, or current pregnancy or breastfeeding status4,15,16.
Ethics Statement
This study was approved by the Human Ethics Committee of Chung Shan Medical University, Taiwan (Approval No. CS1-20035) and the informed consent was confirmed by every involved patient. All methods were performed in accordance with the relevant guidelines and regulations.
Sample Collection
In this study, we collect swabs from the nasopharynx and middle meatus (MM) with rotation at least 5 times under endoscopic guidance. 16 The swabs were placed in a sterile container on ice for immediate transport to the laboratory and were subsequently stored at −80°C until DNA extraction. 17
DNA Extraction
The extraction method followed that of our previous studies15,16. Power fecal pro Kit was used according to the manufacturer’s instructions with additional adding lysozyme (100 mg/ml), at Celsius 37°C for 30 minutes during the sample lysis step. Purified DNA was eluted, quantified, and stored at −20°C.
16S Metagenomics Sequencing
The V3-V4 hypervariable region of 16S rRNA gene was selected and amplified from extracted DNA samples. The PCR primers according to the method in our previous studies using V3 5′-CCTACGGGGNGGCWGCAG-3′ and V4 5′-GACTACHVGGGTATCTAATCC-3′ primers.17,18 Sequences from both ends of 341F-805R primers were trimmed by Cutadapt (v3.5) with the following criteria: read length ≥150°C bp and error rate 0.1 as default.
Microbial Community Analysis and Statistical Analysis
QIIME2 was utilized for the analysis of amplicon sequences, as well as for sample demultiplexing, read denoising, sequence, and sample filtering via feature tables, and estimation of alpha and beta diversity, in addition to taxonomy analysis. For each sample, a minimum of 1000 total reads were obtained and a minimum of 50 total reads were obtained for each feature. Alpha diversity was calculated by implementing the Chao1 diversity index. The Wilcoxon test then compared alpha diversity between the groups. Beta diversity was calculated using the weighted UniFrac distance, and microbiome communities from multiple samples were visualized using principal coordinate analysis (PCoA). SILVA v138 genomic databases were used as a reference for bacteria for taxonomy assignment. To calculate differentially abundant microbiomes (DAMs) between odontogenic and nonodontogenic samples, feature tables with varying taxonomy levels were analyzed using the R package DESeq2. The functional content of the microbiome was inferred based on Operational Taxonomic Units (OTU) using PICRUSt2. 19 STAMP 20 was employed to compute and present the functional pathways along with statistics. Recursive Feature Elimination with Cross-Validation (RFECV) was used for feature selection, while the eXtreme Gradient Boosting (XGBoost) algorithm was utilized to build a predictive model for NPC using DAMs features between NPC and nNPC samples. Model performance was evaluated using the pROC R software package to generate the area under the receiver operating characteristic (ROC) curve (AUC).
Results
Clinical Characteristics of the Patients
This study included consecutive 10 patients with NPC and 15 participants without NPC (non-NPC control group), and Table 1 presents their demographic data. No significant differences with regard to age, gender, asthma, diabetes mellitus, or smoking history were found between these 2 groups.
Demographics of Patients.

Abbreviations: DM, diabetes mellitus; NPC, nasopharyngeal carcinoma.
Use of antibiotics or antifungal medication within 1 month prior to sample collection; Sinonasal or nasopharyngeal surgery.
Undergoing functional endoscopic sinus surgery, septomeatoplasty, skull base surgery, nasopharyngectomy, or adenoidectomy within 1 year prior to sample collection.
Microbial Features and Microbial Diversity of Nasopharyngeal Samples Between NPC and Non-NPC Control Group
Twenty-five nasopharyngeal samples, consisting of 10 cases of NPC and 15 non-NPC controls (nNPC), were subjected to demultiplexing and denoising procedures. On average, each sample had 34,422 reads, which contributed to a dataset consisting of 215 features. Utilizing the SILVA database, these features were then mapped to 58 unique bacterial taxa at the genus level.
However, the nasopharyngeal microbiota showed significant differences and revealed decreased bacterial diversity in NPC groups when compared with the nNPC group. The Chao 1 diversity index in the nasopharyngeal samples of NPC group was significantly lower than that in the nNPC samples (Wilcoxon test P = .021) (Figure 1A).

(A) Shannon diversity (alpha diversity) between microbiomes collected from the nasopharynx in NPC and nNPC groups, as revealed by 16SrRNA sequencing Chao 1 (P = .021). (B) PCoA plot with weighted UniFrac distance. Red and blue dots indicate nasopharyngeal samples from NPC and nNPC groups, respectively. (C) Boxplot of weighted UniFrac distance within each and between groups of NPC and nNPC nasopharyngeal samples. Diversity was significant for weighted UniFrac (P = .001). NPC, nasopharyngeal carcinoma; NP, nasopharynx; nNPC, non-nasopharyngeal carcinoma; PCoA, principal coordinate analysis.
The visualization of PCoA of samples using QIIME2 based on the weighted UniFrac distance was revealed in Figure 1B. Each dot represents an individual bacterial community, incorporating both the presence/absence and relative abundance of bacterial community members, in comparison with all other individuals (closer = more similar; farther apart = more dissimilar). 21 The red and blue dots indicate nasopharyngeal samples from the NPC and nNPC groups, respectively. NPC samples were more closely clustered on the PCoA plot compared to the dispersed nNPC samples.
The weighted UniFrac distances within and between nasopharyngeal samples from the NPC and nNPC groups were analyzed and illustrated (Figure 1C). Permutational multivariate analysis of variance (PERMANOVA) also revealed a significant difference between both groups (pseudo-F = 6.84, P = .001).
The microbiota distribution (relative operational taxonomic unit composition) at the genus level in nasopharyngeal samples from both patients with NPC and nNPC were illustrated. Relative abundance was determined by calculating the ratio of read counts for each feature to the total reads in each sample. The results showed that the microbiota distribution was different between nasopharyngeal samples from patients with NPC and nNPC controls. Pseudomonas was dominant in the samples from the NPC group; by contrast, Streptococcus and Corynebacterium were dominant in the samples from the nNPC group (Figure 2A).

(A) 16S rRNA gene–based bacterial community composition and abundance data for nasopharyngeal samples collected from patients with NPC and nNPC groups. (B) 16S rRNA gene–based bacterial community composition and abundance data for middle meatus samples collected from patients with NPC and nNPC groups. Taxa having less than 1% abundance were annotated as others. NPC, nasopharyngeal carcinoma; NP, nasopharynx; nNPC, non-nasopharyngeal carcinoma; MM, middle meatus.
To identify DAMs of nasopharyngeal samples between NPC and nNPC groups, we filtered genera with a log2-fold change of ≥2 or ≤–2, a P-value of <.05, and a base mean read count of >50 using DEseq2. The genera with the highest differential abundance at the genus level were then summarized and presented in Supplemental Table S1. A positive log2-fold change indicates a higher proportion of a specific bacterium in nasopharyngeal samples from the NPC group compared to the nNPC group, while a negative log2-fold change indicates a lower proportion of the bacterium in the NPC group compared to the nNPC group.
The genera with ≥1% abundance in either group were collected to assess the most prevalent and relatively abundant microbiomes (Figure 3A). Pseudomonas and Corynebacteria had the highest abundance in the nasopharyngeal samples from the NPC group and the nNPC group, respectively. In addition, Paracoccus was rare, occurring most in the NPC group; Neisseriaceae occurred only in the nNPC group.
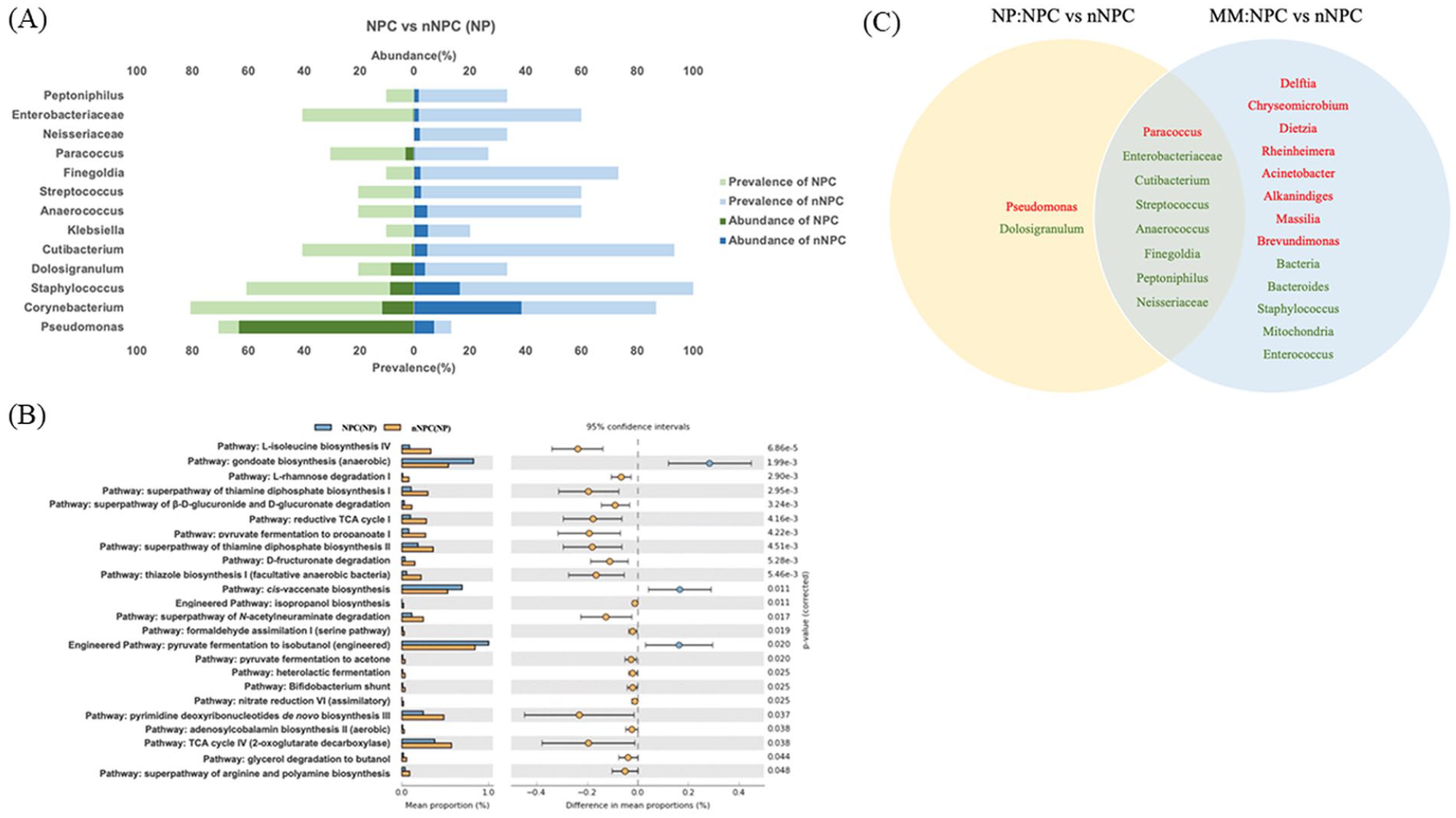
(A) Relative abundance and prevalence based on 16S sequencing data in nasopharyngeal samples from NPC and nNPC groups. Green represents NPC group, and blue represents nNPC group. (B) The functional potential of microbiomes was predicated using 16S rRNA gene sequence data through the application of PICRUSt2 in nasopharyngeal samples of NPC and nNPC groups. (C) Venn diagram of differentially abundance taxa between NPC and nNPC samples collected from the middle meatus and nasopharynx; 8 genera are shown at the intersection. NPC, nasopharyngeal carcinoma; NP, nasopharynx; nNPC, non-nasopharyngeal carcinoma.
The functional analysis of the microbiome between nasopharyngeal samples from NPC and nNPC groups was performed by PICRUSt2. 19 Figure 3B shows that there were significant differences in the abundances of several functional categories between the 2 groups at a false discovery rate of 5%. In the NPC group compared to the nNPC group, there was an increase in the abundance of microbes involved in gondoate biosynthesis (anaerobic), cis-vaccenate biosynthesis, and pyruvate fermentation to isobutanol (engineered) pathway.
Microbial Features and Microbial Diversity of Middle Meatus Samples Between NPC and Non-NPC Control Group
A total of 25 MM samples were demultiplexed and denoised, resulting in a corresponding 16S dataset with an average of 35,150 reads per sample and 248 features. Subsequently, 110 bacterial taxa were annotated at the genus level utilizing the SILVA database.
In contrast to samples from nasopharynx, the samples from the MM showed different features in microbiota. The MM microbiota showed no significant difference in NPC groups when compared with the nNPC group (Wilcoxon test P = .23) (Figure 4A).
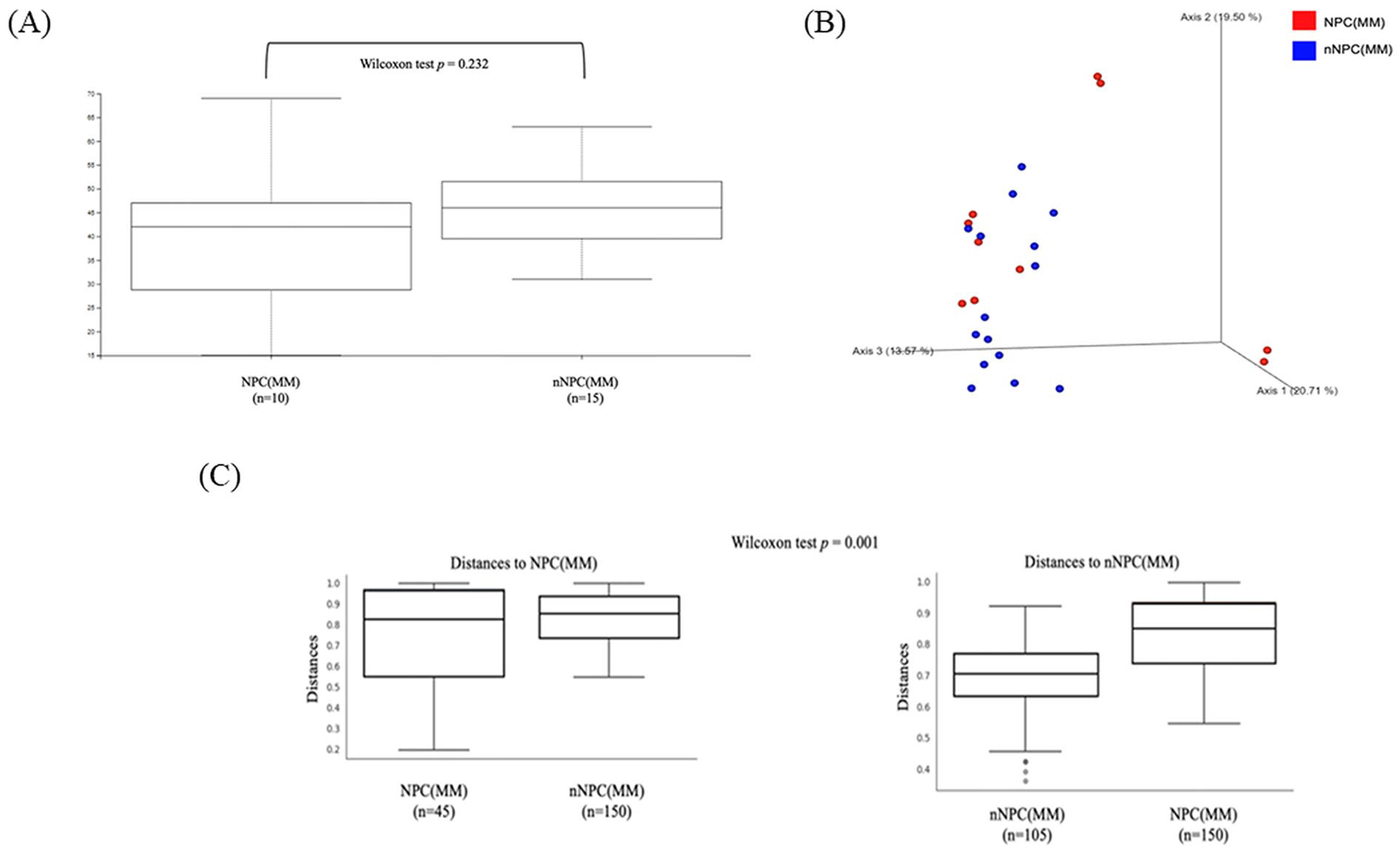
(A) Shannon diversity (alpha diversity) between microbiomes collected from middle meatus in NPC and nNPC groups, as revealed by 16SrRNA sequencing Chao 1 (P = .23). (B) PCoA plot displaying weighted UniFrac beta diversity matrix calculations in middle meatus samples from NPC and nNPC group. (C) Boxplot of weighted UniFrac distance within each and between MM samples from groups of NPC and nNPC. Diversity was significant for weighted UniFrac (P = .001). NPC, nasopharyngeal carcinoma; NP, nasopharynx; nNPC, non-nasopharyngeal carcinoma; MM, middle meatus; PCoA, principal coordinate analysis.
The distribution of microbiota in MM samples of the NPC and nNPC groups is illustrated in Figure 2B. Corynebacterium is the most abundant bacterium found in both NPC and nNPC samples, with a similarity in ratios at 28% and 28.6%, respectively. Dolosigranulum was more abundant in NPC (14.2%) than in nNPC (7.2%), and Staphylococcus was more abundant in nNPC (27%) than in NPC (8%).
Furthermore, MM samples were represented by the visualization of PCoA using QIIME2 based on the weighted UniFrac distance revealed in Figure 4B. In addition, PERMANOVA revealed MM samples from NPC group revealed lower diversity compared with nNPC group (P = .001) (Figure 4C).
The DAMs at the genus level between NPC and nNPC samples in MM were summarized in Supplemental Table S2. A positive log2-fold change indicates a higher proportion of a specific bacterium in MM samples from the NPC group compared to the nNPC group.
The Venn diagram (Figure 3C) demonstrates the intersected DAMs between samples from nasopharynx and MM. The genera labeled in red and green represent higher and lower abundance in NPC group, respectively. There are 8 significant different DAMs present between NPC and nNPC groups both in nasopharyngeal and MM samples, that is, Paracoccus is more abundant in NPC group, while other 7 genera, including Enterobacteriaceae and Cutibeterium, are more abundant in nNPC group.
DAMs in Distinguishing NPC from nNPC
Three bacteria—Pseudomonas, Cutibacterium, and Finegoldia were specifically identified using RFECV from a pool of 10 differentially abundant metagenomics (Supplemental Table S1). Based on these 3 bacteria, a predictive model by machine learning methods was formulated employing an XGBoost approach. Leave-one-out cross-validation showed that the model’s discriminative capacity achieved an AUC-ROC of 0.86, as illustrated in Figure 5A. The Figure 5B heatmap displays the distribution of these 3 bacterial species across the samples. The findings suggest that these bacteria could offer crucial insights into distinguishing between NPC and nNPC samples.

(A) ROC curves of the predictive model-based analysis of differential genera for diagnosis of NPC from nNPC. (B) Heatmap of Pseudomonas, Cutibacterium, and Finegoldia across the nasopharyngeal samples from NPC and nNPC groups. NPC, nasopharyngeal carcinoma; nNPC, non-nasopharyngeal carcinoma; ROC, receiver operating characteristic.
Discussion
This is the first study to analyze commensal microbiome diversity between patients with NPC and control participants without NPC in different locations such as nasopharynx and MM. First, the comparison between nasopharyngeal microbiota from these 2 groups was analyzed. Regarding alpha diversity, the Chao1 diversity index revealed a significant microbiome difference (Wilcoxon test P = .021). PCoA of beta diversity also revealed a significant difference (Wilcoxon test P = .001). The nasopharyngeal samples from NPC group had a significantly high abundance and prevalence of Pseudomonas. Second, among MM samples, although the microbiome diversity was lower in NPC group, there was no significant difference in Chao 1 (P = .232), but beta diversity revealed a significant difference (Wilcoxon test P = .001). Paracoccus was the dominant genus in the NPC group and Staphylococcus was dominant in the nNPC group. Third, after using machine learning methods to analyze the microbiome distribution between NPC and nNPC groups with samples from the nasopharynx, Pseudomonas, Cutibacterium, and Finegoldia can be regarded as the possible biomarker of NPC [area under the curve (AUC): 0.86].
For the first time, we report an association between Pseudomonas and NPC. Pseudomonas aeruginosa is a Gram-negative pathogenic bacterium that presents opportunistic characteristics. 22 Pseudomonas aeruginosa is also associated with cancer, particularly oral squamous cell carcinoma. In a study with 20 fresh oral squamous cell carcinoma (OSCC) cases and aged-gender matched control, Al-hebshi and colleagues found that Pseudomonas aeruginosa is significantly overrepresented in OSCC, while genes involved in bacterial mobility, flagellar assembly, bacterial chemotaxis and lipopolysaccharides (LPS) synthesis were enriched in the tumors. 23 There were also some evidence from in vitro studies to suggest a role in carcinogenesis. 24 Pseudomonas can be isolated from the intestines of cancer patients and has been shown to promote intestinal diseases, including tumorigenesis, when introduced into predisposed model hosts. 24 Morbidity and mortality can be effectively reduced by targeted elimination of Pseudomonas in cancer patients. 24 Elson and colleagues revealed that P. aeruginosa infection caused DNA double-strand breaks in eukaryotic cells, leading to histone H2AX (γH2AX) phosphorylation, repair foci formation in the cell nucleus, and activation of the ATM kinase. 25 These findings imply a possible relationship between P. aeruginosa and cancer biology. 25 Otherwise, the inflammatory response to LPS and flagella, as well as cytotoxins (such as ExoU) possessed by P. aeruginosa, may also play an important role in carcinogenesis due to their potent proinflammatory activity, which leads to the recruitment of neutrophils through the activation of the NF-κB signaling pathway.26,27
In this study, a high association with NPC was first reported for Paracoccus in both nasopharyngeal and MM samples (Figure 3C). Bacteria of the genus Paracoccus inhabit diverse environments, encompassing both pristine and anthropogenically shaped settings. 28 Numerous Paracoccus species hold biotechnological value, with some also being opportunistic human pathogens. 28 Additionally, Paracoccus has been found to be associated with cancer in both human and animal models.29,30 Paracoccus faecalis has been linked to colorectal cancer in animal models. 30 Furthermore, Paracoccus micra, along with other bacteria, was found to be enriched in the gut microbiome of colorectal cancer patients. 29 The potential for Paracoccus to contribute to cancer development has been proposed through mechanisms such as persistent infection, inflammation, DNA damage, initiation of oncogene expression, and immunosuppression. 31 However, the correlation between Paracoccus and malignant tumors requires further evidence and experimental validation in future studies.
Commensal microbiota is the microorganisms that colonize mammals’ mucosal surfaces, with a dynamic community that changes with host evolution and through the lifetime of individual hosts. 32 Intra-tumor microbiota is a community of microbes found in the lining of tumors, affecting patient survival, immune infiltration, and genomic alterations. 33 Commensal microbiota is present on body surfaces covered by epithelial cells and exposed to the external environment, while intra-tumoral microbiota is within the body and activates innate and adaptive immunity. 34 Therefore, the distribution of commensal and intra-tumoral microbiota could be different. In addition to studies on intra-tumoral microbiota, there is limited literature on commensal microbiota. Luo et al collected mucosal samples from the ostiomeatal complex (OMC) of 18 newly diagnosed NPC patients and analyzed the bacterial microbiota using 16S rRNA sequencing, comparing pre- and post-radiotherapy (RT) statuses. 35 Their analysis of pre-RT OMC microbiota identified Corynebacterium as the dominant genus. 35 Our findings similarly demonstrated Corynebacterium as the dominant genus in the MM microbiota. Given the anatomical proximity of the OMC and MM, our results showed consistent outcomes. However, their study did not provide specific information on the pre-RT nasopharyngeal microbiota for direct comparison.
Intriguingly, a comparison of bacterial OTU functional categories using PICRUSt2 19 revealed a notable surge in gondoate biosynthesis among microbes engaged in metabolic pathways within the NPC group (Figure 3B). Notably, previous literature associates gondoate biosynthesis with cancer formation, linking it to pathways such as Dihydroorotate dehydrogenase (DHOD)-driven pyrimidine biosynthesis. 36 The DHODH-driven pyrimidine biosynthesis pathway is crucial in connecting respiration to tumorigenesis, implying a possible connection between gondoate biosynthesis and cancer formation. 36 Moreover, Glycose oxidase has the ability to deplete oxygen levels in tumor microenvironments, leading to heightened acidity, hypoxia, and oxidative stress. 37 These conditions can be exploited for multimodal synergistic cancer therapy. 37 Additionally, gondoate biosynthesis is implicated in cancer through deregulated lipogenesis, altered cancer cell metabolism, and involvement in processes such as hypoxia and oncogene promotion.38,39 The significance of deregulated lipogenesis, inclusive of gondoate biosynthesis, in tumor cell survival underscores its potential as a rational therapeutic target for cancer. 38 Moreover, alterations in cancer cell-intrinsic metabolism suggest a potential role for gondoate biosynthesis in this metabolic reprogramming process. 39
We used RFECV to perform feature selection from 10 DAMs to identify the discriminative features for distinguishing NPC from nNPC. RFECV is a robust feature selection method that uses multiple rounds of cross-validation (10-fold cross-validation repeated 5 times) to increase the reliability of the selected features and improve model performance. The model performance was evaluated using accuracy and kappa metrics, and the highest performance was achieved with 3 selected features (accuracy: 0.7933, kappa: 0.5228). After applying RFECV, the top 3 variables identified were Pseudomonas, Cutibacterium, and Finegoldia. These 3 genera were found to be highly associated with distinguishing NPC from nNPC in our dataset. Their differential presence suggests that they are important biomarkers in the classification task. Therefore, they were selected as the final features for building the predictive model to distinguish between NPC and nNPC groups. While our current model shows good performance, we acknowledge that the relatively small sample size limits the strength of our conclusions. In addition, we recognize that cross-validation alone cannot fully substitute for external validation, especially with a larger cohort. In the future, we plan to increase the sample size to further validate the findings. Our future efforts will include additional recruitment of NPC and non-NPC cases, along with complementary techniques such as PCR and bacterial culture to validate the presence of key bacterial genera identified in the model. These steps will increase the reliability of the model and improve its generalizability, with the ultimate goal of a clinically applicable solution.
This study has several limitations. First, NPC was relatively rare compared with other head and neck cancers; therefore, we were able to include only 10 patients with NPC. Second, fungal microbiota was not included in this study. The evaluation of both fungal microbiota and oral microbiome can enable us to understand the microenvironment between NPC and nNPC groups. Third, we focused on commensal microbiota so intra-tumoral microbiota was not included in this study. Accordingly, future research should include more patients and evaluate the intra-tumoral microbiota, internal transcribed spacers of fungal microbiota as well as oral microbiota to comprehensively elucidate the features of the microbiota of NPC.
Conclusions
To the best of our knowledge, this is the first study to use next-generation sequencing methods to investigate the difference in commensal bacterial microbiota in both nasopharyngeal and MM samples between patients with NPC and those without NPC. Microbiota dysbiosis was significant over the nasopharyngeal samples in patients with NPC compared with those without NPC. Furthermore, Pseudomonas, Cutibacterium, and Finegoldia were potentially served as a diagnostic indicator for NPC.
Supplemental Material
sj-docx-1-ohn-10.1177_19160216241304365 – Supplemental material for Microbial Dysbiosis in Nasopharyngeal Carcinoma: A Pilot Study on Biomarker Potential
Supplemental material, sj-docx-1-ohn-10.1177_19160216241304365 for Microbial Dysbiosis in Nasopharyngeal Carcinoma: A Pilot Study on Biomarker Potential by Yen-Ting Lu, Chung-Han Hsin, Chun-Yi Chuang, Cheng-Chen Huang, Mao-Chang Su, Wei-Sheng Wen, Shao-Hung Wang, Yih-Yuan Chen, Cheng-Yang Lee, Yu-Xuan Li, Ying-Chou Lu, Tzu-Hao Chang and Shun-Fa Yang in Journal of Otolaryngology - Head & Neck Surgery
Supplemental Material
sj-docx-2-ohn-10.1177_19160216241304365 – Supplemental material for Microbial Dysbiosis in Nasopharyngeal Carcinoma: A Pilot Study on Biomarker Potential
Supplemental material, sj-docx-2-ohn-10.1177_19160216241304365 for Microbial Dysbiosis in Nasopharyngeal Carcinoma: A Pilot Study on Biomarker Potential by Yen-Ting Lu, Chung-Han Hsin, Chun-Yi Chuang, Cheng-Chen Huang, Mao-Chang Su, Wei-Sheng Wen, Shao-Hung Wang, Yih-Yuan Chen, Cheng-Yang Lee, Yu-Xuan Li, Ying-Chou Lu, Tzu-Hao Chang and Shun-Fa Yang in Journal of Otolaryngology - Head & Neck Surgery
Footnotes
Acknowledgements
The authors would like to thank Chung Shan Medical University Hospital, Taichung, Taiwan, and St. Martin De Porres Hospital, Chiayi, Taiwan for their assistance with this article.
Availability of Data and Materials
Information regarding the data supporting the results or analyses presented in the article can be reviewed and shared with the journal review committee if needed. However, the authors do not wish to share it publicly owing to their confidential nature.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
This study was performed in accordance with the ethical standards of the institutional and/or national research committee and the guidelines of the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study was approved by the Human Ethics Committee of Chung Shan Medical University, Taiwan (Approval No. CS1-20035) and the informed consent was confirmed by every involved patient.
Supplemental Material
Additional supporting information is available in the online version of the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.