
Abstract
Huntington's disease (HD) is a fatal neurodegenerative disorder characterized by progressive motor, cognitive, and psychiatric symptoms. Research efforts to understand and treat the disease have historically focused on neuronal pathology, but growing evidence underscores the critical role of oligodendrocytes in its pathogenesis. This review synthesizes recent findings on oligodendroglial dysfunction in HD, showing that white matter abnormalities arise early in disease progression, often preceding gray matter changes and clinical symptoms. Neuroimaging and postmortem studies reveal significant white matter atrophy, myelin breakdown, and impaired oligodendrocyte maturation in both patients and animal models. The myelination response to environmental factors is also altered in HD, suggesting impaired white matter plasticity in the disease. At the molecular level, mutant huntingtin disrupts oligodendrocyte function through transcriptional dysregulation of myelin genes, epigenetic modifications involving PRC2 and REST, altered lipid metabolism, thiamine pathway dysfunction, and aberrant BDNF signaling. Key oligodendroglial transcriptional regulators such as MYRF and TCF7L2 are compromised in HD, leading to defective myelination and reduced metabolic support for neurons. Recognizing the role of these mechanisms provides potential biomarkers for early detection and therapeutic targets aimed at preserving both neuronal and glial function in HD.
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat within the HTT gene, leading to an elongated polyglutamine tract in the huntingtin protein. Clinically, HD is characterized by progressive motor dysfunction, cognitive decline, and psychiatric disturbances. Although traditionally considered neuron-centric, emerging research highlights crucial roles for glial cells, particularly oligodendrocytes, in HD pathology.
Oligodendrocytes play an essential role in the central nervous system (CNS), contributing not only to structural support but also to neuronal health and function. First identified in 1921 by Pío del Río Hortega, 1 these cells originate from oligodendrocyte progenitor cells (OPCs), which emerge in multiple waves during embryonic development.2,3 OPCs migrate extensively, guided by chemotactic cues such as growth factors, extracellular matrix components, and axonal guidance molecules. These progenitors ultimately populate the CNS, where they differentiate into mature oligodendrocytes. This process ensures the proper distribution of oligodendrocytes, maintaining CNS functionality throughout life. In adulthood, OPCs retain their proliferative and regenerative potential, albeit at a reduced capacity, which allows for continued production of oligodendrocytes under normal and pathological conditions.4,5
Once differentiated, oligodendrocytes serve a pivotal function in myelinating axons, a process critical for rapid signal conduction. 6 By wrapping axons in layers of myelin, oligodendrocytes enable saltatory conduction, reducing the capacitance of the axonal membrane and dramatically increasing conduction velocity. The structure of myelin itself reflects its functionality, being composed predominantly of lipids and specialized proteins such as myelin basic protein (MBP) and proteolipid protein (PLP). These components not only stabilize the myelin sheath but also influence its ability to insulate axons effectively. Myelin formation is not confined to development; it continues into adulthood, where adaptive myelination and remodeling occur in response to neural activity and environmental stimuli.7,8 This plasticity is essential for cognitive and motor learning, illustrating how dynamic oligodendrocyte function is integral to CNS adaptability.
Oligodendrocytes do more than insulate axons; they provide critical metabolic and trophic support to neurons.9,10 Through the transport of energy substrates such as lactate, oligodendrocytes maintain axonal energy metabolism, ensuring neuronal survival. Additionally, they produce neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF), further underscoring their multifaceted role in sustaining neural networks. The importance of these functions becomes evident in neurodegenerative diseases, where oligodendrocyte dysfunction exacerbates neuronal vulnerability.11,12 Studies in conditions such as multiple sclerosis and leukodystrophies highlight how demyelination and impaired oligodendroglial support contribute to axonal degeneration and clinical decline.
In HD, the role of oligodendrocytes is gaining attention. 13 Recent work suggests that mutant huntingtin (mHTT), the protein responsible for HD, disrupts oligodendrocyte function through multiple mechanisms (detailed below) including transcriptional and epigenetic dysregulation. These disruptions impair the expression of myelin genes, leading to early white matter abnormalities observed in both persons with HD (PwHD) and models of the disease. Such findings emphasize that oligodendrocytes are not passive players in HD but actively contribute to the disease's progression in part by failing to support neuronal health and functionality.
The interplay between oligodendrocytes and neurons is particularly critical in the context of myelin plasticity. This adaptive process allows myelin to remodel in response to changes in neural activity and environmental factors. However, in HD, oligodendroglial plasticity appears compromised,14–17 limiting the capacity to repair and adapt neural circuits. Moreover, mHTT disrupts the metabolic support that oligodendrocytes provide to neurons, further contributing to neuronal damage. These deficits underline the broader implications of oligodendrocyte dysfunction in HD and suggest that targeting this dysfunction may offer new therapeutic avenues.
Emerging research highlights the dynamic roles of oligodendrocytes in regulating neural networks beyond their classical functions in myelination. 18 Their ability to respond to neural activity and environmental changes positions them as central players in CNS homeostasis and adaptability. In HD, understanding how oligodendrocytes contribute to disease pathology opens new possibilities for intervention. By focusing on their transcriptional regulation, myelin production, and metabolic functions, novel strategies may be developed to mitigate neuronal damage and improve clinical outcomes.
Oligodendrocytes are thus indispensable to CNS function, not only through their role in myelination but also as active regulators of neuronal health. Their dysfunction in diseases like HD underscores their importance in maintaining neural integrity. As insights into the biology of oligodendrocytes and their involvement in neurodegeneration deepen, the potential for oligodendroglia-focused therapies grows. These advances hold promise for altering the trajectory of neurodegenerative diseases, including HD, by addressing the critical interplay between glial cells and neurons.
White matter and myelination abnormalities in HD
Although mHTT is expressed in all cells in the human body, neuronal cells in the brain are preferentially affected in HD. Emerging studies indicate that glial dysfunction contributes to neuronal damage in HD, with increasing attention focused on the role of oligodendrocytes in disease pathology (Figure 1). Indeed, oligodendrocyte dysregulation is a common and early pathology in the brains of PwHD, 19 as oligodendrocyte and myelin alterations have been observed in the striatum, corpus callosum, and the fornix of the limbic system of PwHD before onset of motor symptoms.20–24
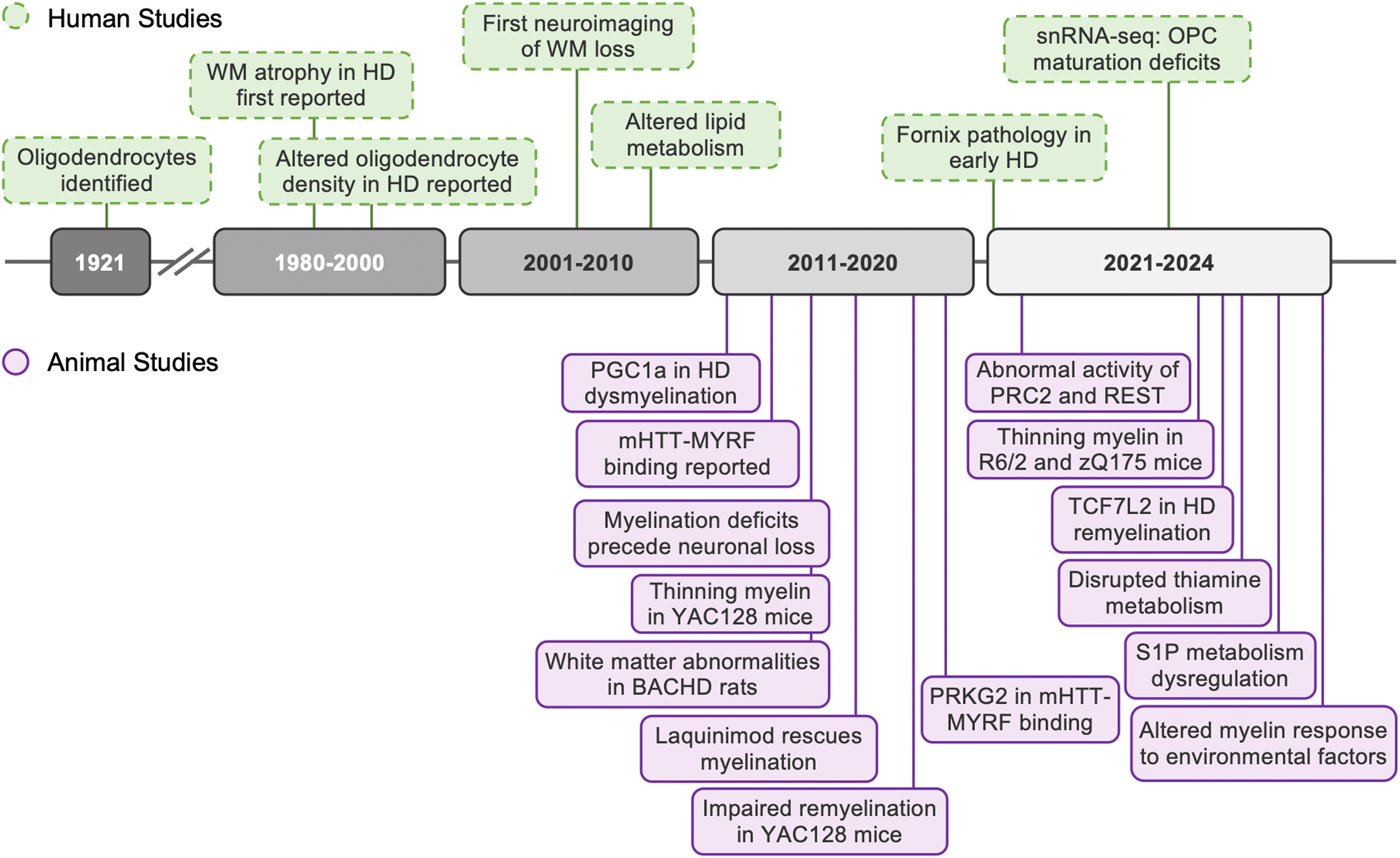
Timeline of notable findings implicating white matter abnormalities and oligodendrocyte pathology in Huntington's disease.
Evidence from human studies
Through neuroimaging studies, McColgan et al. identified white matter loss in PwHD before motor onset, suggesting that this early impairment may result from metabolic dysregulation and axonal terminal damage caused by toxic mHTT.22,23 Changes in white matter including alterations in iron and myelin have been detected as early as 20 years before predicted motor onset in subcortical structures such as the putamen, globus pallidum and the external in the HD-young adult study study. 25 Furthermore, Gabery et al. reported early pathology in the fornix in the limbic system of patients with HD. 24 These comprehensive studies employing volumetric analyses of MRI in the IMAGE-HD study demonstrated that the fornix is smaller in PwHD before motor onset compared to controls. 24 Using postmortem tissue from PwHD, Western blotting analysis confirmed that patients with volume loss of the fornix had lower myelin-regulating factor (MYRF) protein levels compared to non-HD patients. 24 RNA-seq analysis from HD postmortem tissue further provided a possible mechanistic cause for the white matter damage in the fornix. 24 The analysis of transcription factor-target gene interactions demonstrated an enrichment of binding sites for the Polycomb Repressive Complex 2 (PRC2) components on their target genes, as well as the level of RE1 Regulation Transcription Factor (REST). 24 These results suggest that oligodendrocytes in HD suffer from both transcriptional and epigenetic dysregulation at the very early stage induced by mHTT expression directly or indirectly.
Overall, a number of studies of white matter using neuroimaging and analyses of postmortem brain tissue has identified pathology in PwHD. Multiple MRI-based imaging studies in PwHD using both diffusion tensor MRI, neurite orientation dispersion and density imaging (NODDI) and volumetric analyses have demonstrated early and progressive changes in large white matter tracts.26–29 Several studies have demonstrated that white matter loss occurs earlier and progresses more rapidly than gray matter loss in PwHD.30–32 White matter alterations have been detected in several areas of the brain including in the cortico-basal ganglia white matter tracts, the corpus callosum and the limbic system.24,25,33–35
Postmortem analyses of human brain tissue from PwHD have revealed atrophy of white matter 36 as well as increased density of oligodendrocytes in the striatum. 37 A recent study of the white matter tract fornix in the limbic system showed atrophy of this region accompanied by ultrastructural and biochemical evidence of damage to myelin. 24 Bulk and single-cell RNA sequencing of human postmortem tissue from PwHD has shown downregulation of oligodendrocyte identity markers with oligodendrocyte maturation deficits as well as depletion of myelinating oligodendrocytes.24,38,39 Taken together, there is now a wealth of studies using both MRI and postmortem analyses of brains from PwHD that show early and progressive alterations in several white matter areas in HD. These changes include atrophy of the tracts as well as myelin breakdown and disruption of the oligodendrocyte maturation transcriptome.
Evidence from mouse studies
Animal models, particularly rodent models expressing mHTT, have provided crucial insights into oligodendroglial dysfunction in HD (for a detailed review see Ferrari Bardile et al. 13 ). Both fragment and full-length models of HD have shown significant white matter abnormalities. Specifically, R6/2 mice (a fragment model) and zQ175 knock-in mice demonstrate reduced myelin thickness, disrupted axonal myelination, and oligodendroglial transcriptional dysregulation.16,40 Similarly, electron microscopy analyses in YAC128 mice (expressing full-length mHTT) revealed thinning of myelin sheaths.14,17,41,42 These changes parallel the white matter deficits observed in post-mortem HD patient brains. 43
Furthermore, studies using advanced imaging techniques in rodent models of HD have shown reduced fractional anisotropy (FA), indicative of microstructural changes in white matter tracts; e.g., BACHD rats exhibited significantly lower DT-MRI FA values in the anterior corpus callosum, the cingulum and the external capsule at 12 months of age compared with WT littermates. 41 These findings suggest myelination deficits in HD and highlight the potential role of oligodendrocytes. mHTT is expressed in these cells, disrupting their function and leading to impaired myelination and altered neuronal metabolic support. Notably, selective expression of mHTT in oligodendrocytes has been shown to result in progressive motor deficits and reduced lifespan in transgenic mice, along with age-dependent demyelination and decreased expression of myelin genes regulated by myelin regulatory factor (MYRF), a transcriptional activator essential for maintaining myelin gene expression in mature oligodendrocytes. 44 Conversely, inactivating mHTT in oligodendroglia rescues myelin deficits and ameliorates certain behavioral phenotypes in BACHD mice.45,46
Oligodendrocyte progenitor cells (OPCs), which are precursors to mature oligodendrocytes, also exhibit dysfunction in HD models. Reduced proliferation and differentiation of OPCs have been reported, potentially contributing to the observed myelination deficits. In the R6/2 model, OPCs display altered morphology and reduced expression of key markers such as Olig2 and Sox10. 38 Impaired differentiation of OPCs into mature oligodendrocytes results in decreased myelin production and defective repair of demyelinated regions, further exacerbating neuronal vulnerability.
Although this has not been explored in depth to date, the dysfunction of oligodendrocytes in HD is not likely to be solely a cell-autonomous process. Interactions with other glial cells and neurons significantly influence oligodendrocyte health and function. 47 For example, mHTT expression in neurons can alter axonal signaling, impairing the ability of oligodendrocytes to provide effective myelination. Similarly, astrocytes and microglia are known to play critical roles in oligodendrocyte development and function.48–50 Future studies may further clarify these complex astrocyte-microglia-oligodendrocyte interactions and their collective contribution to the myelination abnormalities observed in HD.
Mechanisms of oligodendrocyte dysfunction
Mechanistic studies of white matter pathology in HD have provided insights into how mHTT impacts gene transcription and oligodendrocyte function, implicating several pathways in the pathogenic process (summarized in Figure 2).
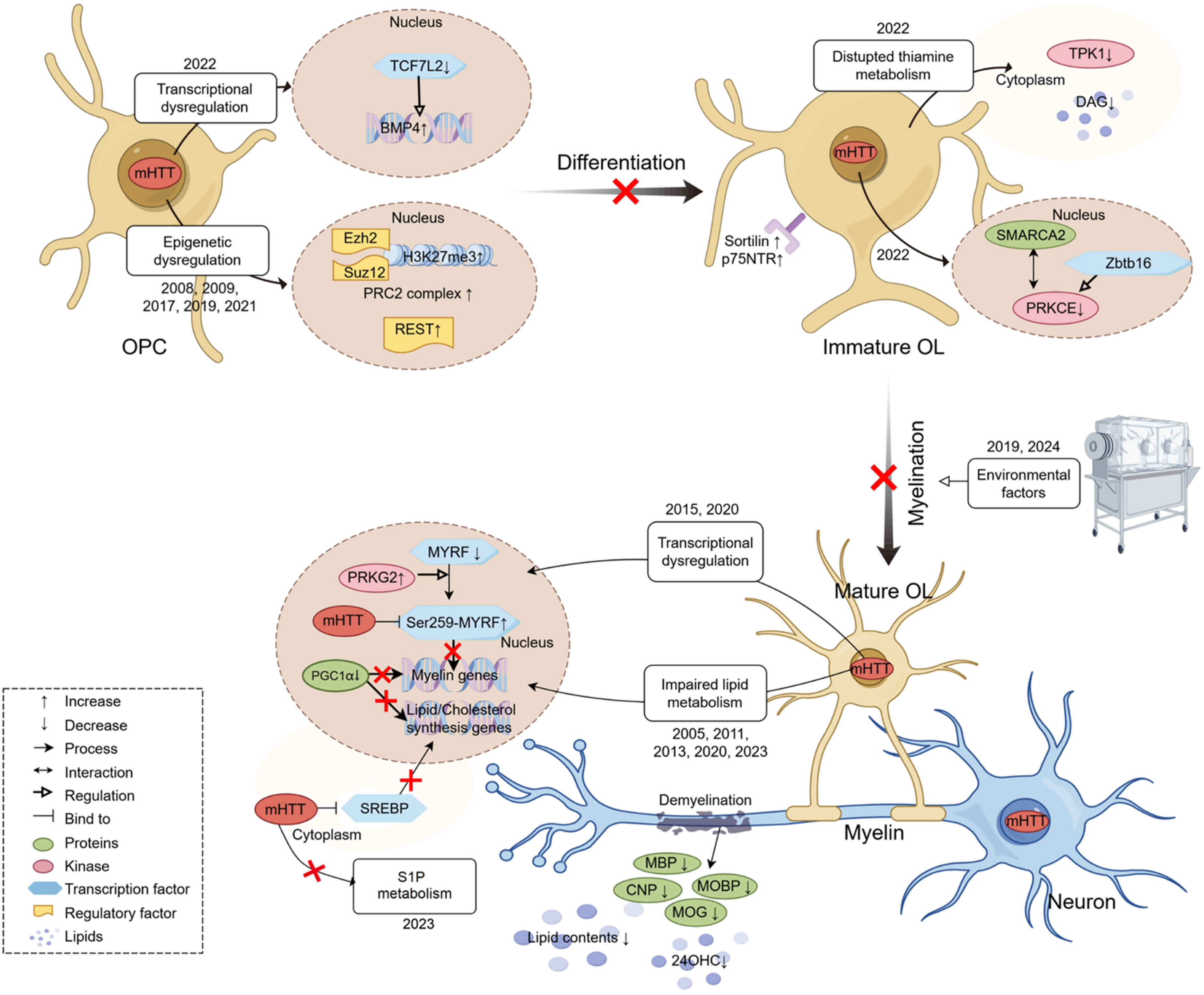
Oligodendrocyte pathogenesis in Huntington's disease. In HD brains, oligodendrocyte pathology arises as early as the stage of oligodendrocyte precursor cells (OPCs) differentiation into immature/mature oligodendrocytes. This process is driven by transcriptional dysregulation, epigenetic remodeling, and disrupted lipid/ sphingosine-1-phosphate (S1P) / thiamine metabolism, and various environmental factors. Ultimately, these alternations lead to myelin-associated protein degradation, lipid loss, and demyelination, impairing neuronal function and integrity. Figure was created using tools on figdraw.com.
Transcriptional dysregulation. One key finding is the association of myelination defects with decreased transcription factor 7-like 2 (TCF7L2)-dependent transcriptional pathways. 16 TCF7L2 plays a crucial role in promoting oligodendrocyte differentiation by tightly regulating bone morphogenetic protein 4 (BMP4) signaling. Reduced TCF7L2 levels lead to increased BMP4 expression in oligodendrocytes, inhibiting the generation and differentiation of oligodendrocytes. 51
In a mouse model expressing the N-terminal 251 amino acids of mHTT driven by the PLP-promoter (PLP-150Q HD mice), demyelination was observed at 3–5 months. 44 The demyelination in PLP-150Q mice was attributed to mHTT's abnormal binding to MYRF, affecting its transcriptional activity and reducing the transcription of myelin genes, including MBP, 2′,3′-Cyclic nucleotide 3′-phosphodiesterase (CNP), myelin-associated oligodendrocyte basic protein (MOBP), and myelin oligodendrocyte glycoprotein (MOG). 44
A subsequent study using laquinimod (LAQ) to treat PLP-150Q HD mice significantly reduced myelin abnormalities by decreasing cGMP-activated protein kinase subunit II (PRKG2). Knocking down PRKG2 using CRISPR/Cas9 further confirmed that abnormal PRKG2 activity enhances mHTT binding to MYRF, affecting its function. 52 Reduced PRKG2 levels lead to a decrease in phosphorylated Ser259-MYRF, releasing MYRF from the mHTT bond and restoring its normal activity. 52 The myelin repair effect of LAQ was also demonstrated in YAC128 mice, showing independence from inflammatory regulation, as LAQ acted by regulating inflammation in the treatment of multiple sclerosis. 42
Furthermore, attenuating mHTT's effects on peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), a key regulator of MBP production and cholesterol synthesis in oligodendrocytes, may ameliorate early myelin dysregulation.53,54 These studies shed light on the molecular mechanisms underlying white matter damage in HD and suggest potential therapeutic targets for mitigating oligodendrocyte dysfunction and myelin abnormalities in the disease.
Altered lipid metabolism. In addition to the mechanisms previously discussed, impaired myelin dysregulation in HD can also be influenced by altered lipid metabolism. Myelin, a lipid-enriched and highly organized multi-layer membrane structure, plays a crucial role in facilitating fast, long-distance saltatory conduction of neuronal impulses. 55 Animal brain myelination and remyelination rely on fatty acid synthesis in oligodendrocytes. 56 Several investigators have reported altered lipid metabolism in the brains of individuals with HD.57,58
The impaired lipid metabolism in HD may result from reduced biogenesis due to decreased levels of brain-specific catabolite 24S-hydroxycholesterol (24OHC) or due to the binding of mHTT to SREBP, inhibiting its translocation to the nucleus and activating cholesterol and lipid biosynthesis.59–61 For example, in 9-month-old LacQ140 HD mice, a decrease in overall striatal lipid content was observed, driven primarily by a reduction in subclasses crucial for white matter integrity, such as ceramide, sphingomyelin, and monogalactosyldiacylglycerol. 62 Alterations were also noted in phosphatidylinositol, phosphatidylserine, and bismethyl phosphatidic acid. 62 Transcriptional changes affecting myelin lipids showed partial reversal with reduction of mHTT levels. 62
Research by Pepe et al. demonstrated that the inhibitor 2-acetyl-5-tetrahydroxybutyl imidazole (THI), an inhibitor of the sphingosine-1-phosphate (S1P) degradative enzyme SGPL1, significantly reduces myelin damage in HD and restores myelin marker proteins in HD models, supporting the dysregulated lipid or fatty acid mechanism in HD.63,64 These findings highlight the importance of lipid metabolism in myelin integrity and suggest potential therapeutic targets for addressing myelin impairment in HD through modulation of lipid pathways.
Epigenetic mechanisms. Epigenetic dysregulation appears to be another critical mechanism contributing to oligodendrocyte dysfunction in HD. Studies in both human and murine HD models have identified abnormal activity of key epigenetic regulators, including the PRC2 and repressor element-1 silencing transcription factor (REST).24,45 Of particular interest is the interaction between mHTT and PRC2, previously shown to enhance PRC2’s histone H3K27 trimethylase activity, 65 which is reflected in increased H3K27me3 levels in the corpus callosum of BACHD mice. 45 This heightened PRC2 activity disrupts the transcriptional regulation of myelin-related genes, as evidenced by the increased binding of PRC2 subunits to their regulatory regions. 45 Key components of the PRC2 complex, such as enhancer of zeste homolog 2 (Ezh2) and suppressor of zeste 12 (Suz12), are essential for OPC differentiation into mature oligodendrocytes by silencing progenitor-specific gene programs.66,67 Notably, selective inactivation of mHTT in oligodendroglia of BACHD mice restored normal patterns of Ezh2 and Suz12 binding, further implicating this pathway in oligodendrocyte dysfunction in HD and highlighting its potential as a therapeutic target. 45
Role of BDNF and environmental factors. Although mature BDNF plays a significant neuroprotective role, 68 its precursors are believed to cause synaptic depression and cell death.69,70 A study showed that immature oligodendrocytes in the striatum exhibit markedly elevated levels of the BDNF precursor receptors sortilin and p75NTR, which are believed to be closely associated with HD striatal myelin degradation. 71
Two recent studies demonstrated that the environment can also influence oligodendrocyte pathology in HD.15,17 In a germ-free (GF) environment, mature oligodendrocytes and myelin-associated proteins were reduced in the prefrontal cortex in both wild-type (WT) and BACHD mice. Intriguingly, ultrastructural studies revealed that the GF environment exerted a more pronounced effect on myelin and axons in BACHD mice. 15 Environmental deprivation experiments demonstrated that myelin sheaths of YAC128 mice were less affected compared to those of wild-type (WT) mice under impoverished housing conditions, suggesting a paradoxical resistance of YAC128 mice to adverse environmental conditions. 17
PRKCE and thiamine metabolism. Recent studies have identified protein kinase C epsilon (PRKCE) and thiamine metabolism as important regulators of oligodendrocyte maturation in HD. 38 PRKCE, regulated by diacylglycerol (DAG) and the transcriptional factor Zbtb16, interacts with the chromatin remodeler SMARCA2 to influence OPC differentiation. In HD brains and R6/2 mice, reduced PRKCE and DAG levels disrupt oligodendrocyte maturation. 38 Thiamine metabolism also appears to contribute to oligodendrocyte dysfunction in HD. Thiamine pyrophosphokinase 1 (TPK1) is dysregulated in R6/2 mice, affecting acetyl-CoA and DAG production. Notably, thiamine and biotin supplementation rescued oligodendrocyte maturation deficits in R6/1 HD mice, further supporting a role for this pathway in oligodendrocyte dysfunction in HD. 38
Conclusions
Oligodendrocytes contribute significantly to HD pathogenesis through mechanisms extending beyond their classical role in myelination. As evidenced by neuroimaging studies, postmortem analyses, and preclinical models, white matter abnormalities emerge early in disease progression, often preceding gray matter changes. The molecular underpinnings of oligodendrocyte dysfunction in HD involve mHTT-mediated disruption of key transcriptional regulators such as MYRF and TCF7L2, dysregulated lipid and thiamine metabolism, altered BDNF signaling, and epigenetic dysregulation. These mechanisms collectively impair oligodendrocyte differentiation, myelin production, and metabolic support for neurons, exacerbating neuronal vulnerability and disease progression. These molecular disruptions are compounded by the pro-inflammatory environment created by interactions with astrocytes and microglia, which further impair oligodendrocyte health and function.
By recognizing HD as a disease with substantial oligodendroglial involvement, novel therapeutic strategies targeting white matter integrity and oligodendrocyte function can be developed. However, critical knowledge gaps remain. Future research should address the temporal relationship between oligodendrocyte dysfunction and neuronal pathology, regional heterogeneity in oligodendroglial responses, and whether oligodendrocyte-specific interventions can modify disease progression. Furthermore, the interplay between genetic factors and environmental influences on oligodendrocyte health in HD requires further investigation.
Understanding the mechanisms of oligodendrocyte dysfunction in HD may reveal biomarkers for early detection and therapeutic monitoring, potentially leading to combinatorial approaches targeting both neuronal and glial pathologies to effectively alter disease trajectory in PwHD.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported grants to MAP. from the BC Children's Health Research Institute (IGAP, Investigator Grant Award), Michael Smith Health Research BC (Scholar Award SCH-2020-0656), and the Huntington Society of Canada (AWD-019707); research grants to A.P. from the Swedish Research Council (grant number 2022/01092), the Swedish Brain Foundation, the Swedish governmental funding of clinical research (ALF) at Region Skåne, and the Knut and Alice Wallenberg Foundation (#2019.0467); research grants to H.P.N. from the German Research Foundation (DFG grant number NG101/6-1) and the Federal Ministry of Education and Research (BMBF, grant number: 01ED2406); grants to X.-J.L. and S.L. from the National Key R&D Program of China (2021YFA0805200), the National Science Foundation of China (82271902, 82071421, 82394422, 82371874, 81830032, 31872779, 81922026, 82171244), and the Guangzhou Key Research Program on Brain Science (202007030008, 202007030003).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: HPN, AP, and MAP are Editorial Board Members of this journal but were not involved in the peer-review process of this article nor had access to any information regarding its peer-review.