
Abstract
Background
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder characterized by parkinsonism, cerebellar ataxia, and autonomic dysfunction. Putaminal hypointensities on T2 magnetic resonance imaging (MRI) are commonly observed in the parkinsonian variant of MSA (MSA-P).
Objective
To report a neuropathologically confirmed case of MSA-P with an atypical MRI presentation, characterized by progressive T2 hyperintensity in the putamen, without significant iron accumulation.
Methods
We present the clinical course, imaging findings, and neuropathological results of a 57-year-old woman with MSA-P. Diagnostic evaluations included serial brain MRI, cerebrospinal fluid analysis, magnetic resonance spectroscopy, and post-mortem pathological examination.
Results
The patient initially presented with L-dopa-responsive bradykinesia and right-dominant rigidity, followed by progressive motor decline, orthostatic hypotension, and urinary retention. Serial T2-weighted MRI revealed diffuse and progressively increasing hyperintensity in the putamen, accompanied by atrophy predominantly on the left side. Autopsy confirmed the diagnosis of MSA, revealing severe neuronal loss, marked gliosis, and abundant glial cytoplasmic inclusions in the putamen, with only mild iron deposition as shown by Prussian blue staining.
Conclusions
This case demonstrates that severe putaminal neurodegeneration in advanced MSA can be associated with progressive T2 hyperintensity on MRI, reflected by the absence of substantial iron deposition. These findings indicate that iron-independent mechanisms contribute to the pathophysiology of MSA-related putaminal damage in patients with MSA.
Plain language summary
Multiple system atrophy (MSA) is a rare and progressive brain disease that affects movement and autonomic functions, such as blood pressure control and bladder function. Brain imaging plays a significant role in establishing the diagnosis, particularly through changes in increased signal intensities in the putamen region on magnetic resonance imaging (MRI), which reflect neurodegeneration and iron accumulation. Here, we describe a woman in her late 50 s who developed MSA with motor symptoms, such as slowness and stiffness, along with later problems such as orthostatic hypotension and urinary difficulties. Her brain scans revealed unusual findings: instead of the typical hypointense areas associated with iron accumulation, the putamen appeared hyperintense, with the signal intensity progressively increasing over time. Several years later, autopsy revealed severe damage to the putamen, with only a small amount of iron remaining. This case illustrates that significant brain damage can occur without substantial iron accumulation in MSA, suggesting that iron-independent mechanisms may play important roles in disease progression. Understanding these processes could help improve the diagnosis and treatment of MSA.
Introduction
Multiple system atrophy (MSA) is a type of α-synucleinopathy that primarily presents with parkinsonism, cerebellar ataxia, and autonomic dysfunction. 1 While no curative treatments are currently available for MSA, symptom management, including orthostatic hypotension and urinary disorders, can significantly improve activities of daily living (ADL). Pathologically, MSA is characterized by glial cytoplasmic inclusions (GCIs) in oligodendrocytes. 2 Neuronal loss predominantly affects dopaminergic neurons, with additional involvement of the putamen, caudate nucleus, and myelinated fibers of the basal ganglia. These findings are reflected in the MRI results showing slit-like hyperintensity on the lateral edge of the putamen, iron deposits in the dorsal putamen in T2*, the “hot cross bun” sign in the pons, and the middle cerebellar peduncle (MCP) sign, all of which are now included in diagnostic criteria. 3 Here, we present a case of MSA with distinctive MRI findings, demonstrating extensive high-intensity signals throughout the entire putamen without accompanying linear changes along its borders.
Case report
A female patient developed bradykinesia and right-dominant rigidity at the age of 57. Initially, her symptoms responded well to L-dopa. However, after two years of relatively slow progression during which she had been able to walk independently, her motor symptoms progressively deteriorated over several months. Despite increasing the dose of L-dopa/carbidopa to 850 mg daily and administrating entacapone at 300 mg daily, she eventually required a wheelchair for most indoor activities and relied on furniture or walls for support when walking to the bathroom. At the initial visit to our hospital, the UPDRS-III score was approximately 45, and no other anti-Parkinsonian drugs were administered. Cognitive function and autonomic function were relatively preserved. There was no evident signs of cerebellar ataxia or REM sleep behavior disorder. However, the early manifestation of dropped head syndrome was not inconsistent with a diagnosis of MSA, although its response to levodopa was minimal. Orthostatic hypotension was not evident in the early stage but developed as the disease progressed. Similarly, urinary retention became apparent in the advanced stage. In November 2017, at age 60, a brain MRI revealed diffuse mild hyperintensity in the putamen on T2-weighted images and isointensity on T1-weighted images (Figure 1(a)). Around two years after, in December 2019, at age 62, the hyperintensity became more pronounced on T2-weighted images, accompanied by putaminal atrophy predominantly on the left side, but without hyperintense rim (Figure 1(b)). Correspondingly, the atrophic area showed hypointensity predominantly on the left side on T1-weighted images (Figure 1(b)). Differential diagnoses, including mitochondrial disease, primary central nervous system malignant lymphoma, bilateral hypoxic encephalopathy, osmotic myelitis, metabolic or drug-induced conditions, all of which can cause putaminal hyperintensity, were excluded through cerebrospinal fluid (CSF) and magnetic resonance spectroscopy (MRS) examinations. CSF analysis revealed no pleocytosis and oligoclonal bands were negative. There was no apparent elevation in the lactate-to-pyruvate ratio. Microbiological cultures were negative for infectious pathogens, and autoimmune panel was not submitted. Magnetic resonance spectroscopy of the brain demonstrated a mild reduction of N-acetylaspartate (NAA) in the lesion in the left putamen, relative to normal white matter, which was considered consistent with a diagnosis of MSA. At the age of 63, she was tentatively diagnosed with multiple system atrophy of the parkinsonian type (MSA-P) based on L-dopa unresponsive parkinsonism and orthostatic hypotension. 3 By the age of 64, the patient developed dysphagia accompanied by intermittent choking episodes. The emergence of worsening of orthostatic hypotension along with progressive limb weakness further contributed to her functional decline, ultimately rendering her bedridden. The putaminal hyperintensity on T2-weighted images became more prominent and distinctly delineated, but no hypointensity was observed on T2*-weighted images (Figure 1(c)). However, on T1-weighted images, the area, particularly on the left side, appeared to be slightly isointense (Figure 1(c)). Despite undergoing gastrostomy, she died at age of 64 due to pneumonia. All brain MRI examinations were performed using a 1.5 Tesla-scanner; however, susceptibility-weighted imaging was not included. All images were reviewed by a neuroradiological specialist certified by Japan Radiological Society.
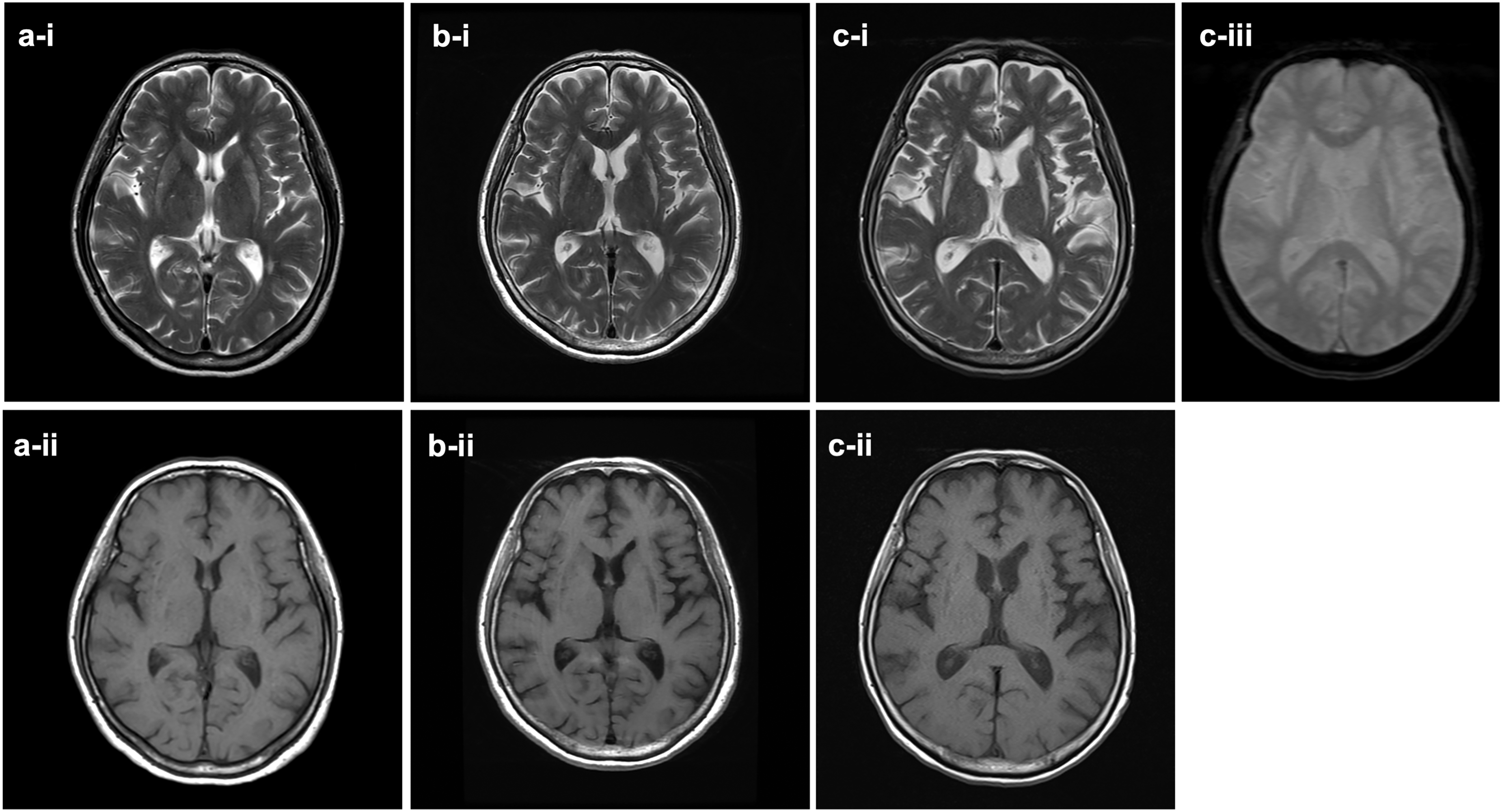
Brain MRI shows progressively increased hyperintensity in the bilateral putamina on T2-weighted images without an accompanying hyperintense rim surrounding the putamen (a-i, b-i, c-i). The putamina shows gradual putaminal hypointensity on T1-weighted images (a-ii, b-ii, c-ii) but not hypointensity on T2* images (c-iii). These MRI imaging were performed on November 17, 2017 (a), December 2019 (b), and December 2022 (c).
Macroscopically, there was severe putaminal atrophy with spongiform changes observed throughout the rostral to caudal regions. Marked depigmentation was noted in the substantia nigra and locus coeruleus. Degenerative changes were predominantly observed in the nigrostriatal system, aligning with characteristic MSA-P. Severe neuronal loss with gliosis was microscopically observed throughout the entire putamen. The extent of neurodegeneration in other brain regions was generally consistent with that observed in typical cases of MSA-P. Abundant glial cytoplasmic inclusions (GCIs) were detected in both the nigrostriatal and olivopontocerebellar system, as well as in the cerebral white matter. Perls’ Prussian Blue staining demonstrated only mild iron deposition in the putaminal rim, which contrasts with the more pronounced iron deposition typically observed in MSA (Figure 2(a)-(d)). All pathological findings were reviewed by a certified neuropathologist of the Japanese Society of Neuropathology.
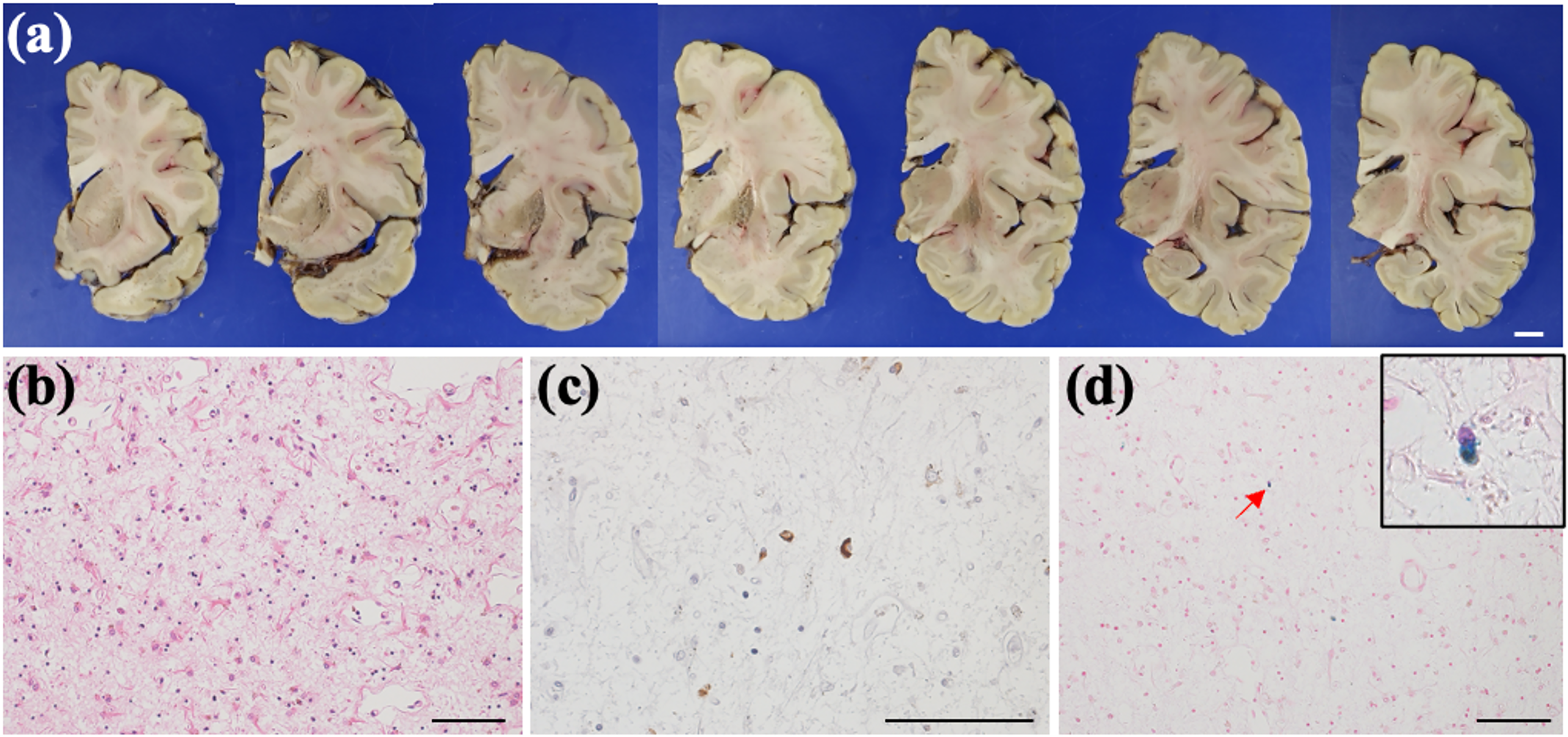
The post-mortem brain section exhibits severe putaminal atrophy (a), marked by neuropil rarefaction accompanied by significant neuronal loss and gliosis (b). Furthermore, glial cytoplasmic inclusions are evident (c), while no signs of iron accumulation are observed (d). Hematoxylin and eosin (b), anti-phospho-α-synuclein (c), and Berlin blue staining (d) are utilized. Scale bars are 1 cm (a) and 100 µm (b-d).
Discussion
This case was characterized by distinctive MRI findings of progressive putaminal hyperintensity on T2-weighted imaging. The diagnosis of MSA was eventually confirmed pathologically. MRI imaging features of the MSA-P have been attracted much attention because of their contribution to establishing the diagnosis. Initially, hyperintense lesions around putamen was considered important. 4 However, subsequent findings revealed that these lesions are less specific to MSA, as similar manifestations can occur in Parkinson's disease. 5 More recent observations indicate such lesions can even appear in healthy individuals depending on the MRI sequence used. 6 Another important diagnostic feature is putaminal atrophy, characterized by a reduction in putaminal volume and is considered valuable because of its specificity to MSA. In addition to size reduction, signal changes within the putamen, characterized by the decreased signal intensity on T2-weighted images and diffusion tensor imaging, likely due to iron deposition, are also important imaging features. 7
In this case, from the early stages of the disease, there was a widespread increase in putaminal high signal intensity on T2-weighted images, which prompted consideration of differential diagnoses such as metabolic, malignant, or drug-induced conditions. While rapid progression led to overall atrophy of the putamen, consistent with previous findings, the absence of significant iron deposition was notable. Additional, minimal hyperintense putaminal rim was a distinctive feature in this case.
MSA affects various brain cell populations, leading to degeneration across multiple systems. It is characterized by widespread GCIs associated with neurodegenerative changes in structures such as the striatonigral system. 2 GCIs are found in demyelinated white matter tracts as well as in the basal ganglia, brainstem, cerebellum, and motor cortex, correlating with neuronal loss. MSA-P typically begins with minimal change in the substantia nigra and progresses to the putamen, caudate, and globus pallidus. In this case, neurodegeneration with GCIs that was most pronounced in the nigrostriatal system, which is pathologically consistent with MSA-P. When comparing the putaminal change to external capsule, which showed minimal imaging changes, we observed severe gliosis in the putamen (Figure 2(a)), reflecting MRI findings.
MSA is also characterized by the pathological feature of the iron deposition, which is crucial for myelination in the developing brain and is primarily located in oligodendrocytes in iron-rich brain regions, particularly in the degenerated areas. The iron accumulation in the putamen may be associated with increased oxidative stress and α-synuclein aggregation in MSA. 8 However, the exact mechanism behind this and whether iron promotes neurodegeneration or results from it remain unclear. The putamen in MSA-P contains the highest level of intracellular iron deposition, as observed by Perl's staining across several brain regions in both MSA-P and MSA-C. 9 The percentage of putaminal iron deposition reached 80% in MSA-P, which is higher than that in PD and healthy controls, as observed by susceptibility-weighted imaging. 10 Among the cell types with iron deposition in the putamen, microglia exhibit the most significant accumulation of iron. By phagocytosing iron, microglia may contribute to ferroptosis by generating reactive oxidative stress, which can lead to neurotoxicity. 9 In contrast, in the current case, despite severe gliosis in putamina, iron deposition was markedly minimal compared to typical MSA cases (Figure 2(a)-(d)). This suggests that iron deposition contributed minimally to the neuronal loss observed in this case. In MSA pathological iron accumulation is generally correlated with disease progression and neurodegeneration, as demonstrated by both histopathological analysis and MRI-based studies. 11 However, certain brain regions, such as the locus coeruleus, exhibit severe neuronal loss despite minimal or absent iron deposition, suggesting that iron-independent mechanisms may also contribute to neurodegeneration in MSA. Previous reports have proposed that GCIs, composed of misfolded α-synuclein, may drive neurodegeneration through proteotoxic stress, microglial activation, and the release of proinflammatory cytokines, even in the absence of iron-induced oxidative damage. These observations suggest that although iron accumulation may exacerbate disease progression, it is not a prerequisite for neurodegeneration in all cases of MSA. Based on these findings, we hypothesize that iron-independent mechanisms may also contribute to the neurodegenerative process in MSA.
We report a case of MSA, characterized by MRI findings of putaminal extensive hyperintensity on T2-weighted images. These imaging findings correlated with the underlying pathological process of severe gliosis, which predominantly observed in the putamen compared to other brain lesions. In more advanced cases of MSA, extensive putaminal changes on MRI may reflect severe neurodegeneration.12,13 This case offers valuable insight on the relationship between brain iron homeostasis and neurodegeneration, warranting further investigation into MSA pathophysiology.
Footnotes
Ethical considerations
Written informed consent was obtained from subjects participating in this study, according to the Declaration of Helsinki. The study was approved by the Ethics Committee of the Juntendo University School of Medicine (No. 2018095). We confirmed that we have read the Journal's position on issues involved in ethical publication and affirmed that this work is consistent with those guidelines.
Consent to participate
All patients included in this study provided informed consent for their information to be used.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Ueno, Dr Tsunemi, and Dr Taniguchi have no conflict of interest to report. Prof. Hattori is an Editorial Board member of this journal, but was not involved in the peer-review process of this article nor had access to any information regarding its peer review.
Data availability statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.