
Abstract
Background
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by the progressive loss of dopaminergic neurons. While abnormal protein aggregation has been classically implicated in PD, increasing evidence suggests that lipid dysregulation may also contribute to neuronal vulnerability. Recent studies have begun to link abnormal phosphatidylserine (PS) metabolism to mitochondrial impairment and dopaminergic neuron loss in PD, yet the underlying cellular mechanisms remain poorly defined.
Objective
This study aimed to determine how impaired PS synthesis in cortex glia affects mitochondrial function, oxidative stress, and dopaminergic neuron survival, using a Drosophila model of glia-specific Phosphatidylserine synthase (Pss) knockdown.
Methods
To dissect the glial contribution to PS-related neurodegeneration, we employed a Drosophila model in which the Pss gene was selectively knocked down in cortex glia using the GAL4-UAS system. We evaluated PD-like phenotypes by assessing the number of dopaminergic neurons in the PPL1 and PPL2 clusters, as well as locomotor activity and lifespan, following glia-specific knockdown of Pss gene.
Results
Cortex glia-specific knockdown of Pss impaired locomotion and reduced lifespan in flies, indicating a systemic decline in neuronal and mitochondrial function. Pss knockdown reduced mitochondrial transcription factor A (Tfam) expression, disrupted mitochondrial gene expression, and elevated ROS levels. Western blot analysis also revealed reduced AKT phosphorylation without changes in total AKT. These results ultimately lead to loss of dopaminergic neurons.
Conclusions
These findings establish a mechanistic link among abnormal PS metabolism, impaired AKT signaling, mitochondrial dysfunction, and dopaminergic neuron loss. Our study provides novel evidence that glia-driven abnormalities in PS metabolism may cause PD-like neurodegeneration, offering mechanistic insights and potential therapeutic targets.
Plain language summary
Parkinson's disease (PD) is a neurodegenerative disorder that affects millions of people worldwide. In PD, certain brain cells that produce dopamine slowly die, partly because of harmful protein buildup. More recently, scientists have found that changes in certain brain fats, called phospholipids, may also play a role in PD. However, how these changes lead to brain cell damage remains unclear. In this study, we focused on a specific type of phospholipid called phosphatidylserine (PS). PS is an important lipid that helps maintain brain cell health, particularly by supporting the function of their energy centers, known as mitochondria. We aimed to determine that abnormal PS metabolism could harm brain cells. To study this, we used fruit flies, which share many biological similarities with humans. We examined glial cells in the fly brain—these are support cells that help protect and maintain neurons. When we reduced the activity of the gene responsible for producing PS in these glial cells, (phosphatidylserine synthase; Pss), the flies lost dopamine-producing neurons. The flies also moved less and had shorter lifespans. Additionally, reducing PS production impaired mitochondrial function in glial cells. This led to a buildup of harmful molecules called reactive oxygen species, which damaged nearby nerve cells. PS loss affected how mitochondria produce energy and manage stress. These findings suggest that when brain cells cannot produce enough PS, their energy systems begin to fail, leading to neuronal loss. This may be one of the ways that PD begins in the brain. Understanding this process could inspire new treatment approaches in the future.
Introduction
Parkinson's disease (PD) is a neurodegenerative disease common worldwide. PD involves the gradual death of cells in multiple parts of the brain.1,2 Death of dopaminergic neurons in the substantia nigra, located in the midbrain, reduces dopamine levels in the nervous system. A decrease in dopamine, which is an essential signal for coordinating movement in the brain, leads to motor abnormalities such as stiffness, shaking, and difficulty with balance.3,4 Until recently, the cause of PD remained largely unknown.
The pathogenesis of PD remains unknown because only 10% of patients have Mendelian PD. Most PD patients are classified as sporadic. 5 Therefore, the causes of PD are poorly understood. Gene mutations and environmental toxins are associated with the development of PD. In cases of Mendelian PD, the dysfunction of PINK1/PARKIN may result in early-onset PD. 6 PINK1 or PARKIN mutations lead to impaired mitochondrial quality control, affecting the process of clearing damaged mitochondria, which is called mitophagy. 7 Mitophagy is vital for preventing cell damage by removing dysfunctional mitochondria. Mitochondrial dysfunction is a crucial factor in the pathogenesis of PD, and genes involved in mitochondrial quality control play a significant role in disease susceptibility. Recent studies have indicated that certain phospholipid synthases, particularly phosphatidylserine synthase (PSS), may play critical roles in mitochondrial quality control, resulting in neurodegeneration and muscle atrophy.8,9
Pss is a phosphatidylserine synthase. In the brain, Pss appears to be expressed in glia but not in neurons. 9 Our and other groups have shown that Pss plays a critical role in neurodegeneration and muscle atrophy.8,9 Although Pss is an essential gene for development, our study recently uncovered its pathological roles in adulthood. Decreased Pss expression has been implicated to play a role in the pathogenesis of the autism spectrum disorder. We demonstrated that in flies, reduced Pss expression results in mitochondrial dysfunction and increased reactive oxygen species (ROS) production, leading to the development of neurodegeneration and muscle atrophy.8,9
In sporadic PD, genome-wide gene expression profiles of the brain have revealed altered networks in mitochondrial and autophagic systems. 10 In addition, genome-wide association studies have found multiple loci linked to risk, suggesting that unknown genetic factors, together with environmental toxins, may be the causal elements of sporadic PD.11,12 Identifying significant elements within the pathways involved in sporadic PD is essential to prevent rapid neurodegeneration. Given that Pss is crucial for mitochondrial integrity and cell death in glia, particularly cortex glia, we hypothesized that Pss downregulation in cortex glia may contribute to PD pathogenesis. 9 We utilized fly models with Pss knockdown specifically in the cortex glia to investigate its function in cortex glia. We found that Pss downregulation in cortex glia causes mitochondrial dysfunction and dopaminergic neuronal death, demonstrating that phosphatidylserine in cortex glia could be an important factor in PD.
Methods
Drosophila genetics and strains
Canton-S and UAS-Dcr2 (#24646) flies were obtained from the Bloomington Drosophila Stock Center (Indiana University, Bloomington, IN, USA), NP2222-GAL4 (#112830) flies were obtained from the Kyoto Drosophila Stock Center (Kyoto Institute of Technology, Kyoto, Japan), and UAS-Pss RNAi (#v5391) flies were obtained from the Vienna Drosophila Resource Center (Vienna, Austria). UAS-Pss (II) has been described previously. 9 We used the NP2222-GAL4 line to collect males for crossing with females of UAS-Dcr2; UAS-Pss RNAi (NP2222 > Dcr2, Pss RNAi, for knockdown [KD]) or UAS-Pss (NP2222 > Pss, for overexpression [OE]). The flies were raised in vials containing standard cornmeal medium. Only F1 males were used for counts in the experiments that required adults. Fly culture, crossings, and analyses were performed using standard fly medium at 25 °C with 50% relative humidity and a 12 h: 12 h light: dark cycle.
Reverse transcription quantitative real-time PCR (qrt-PCR)
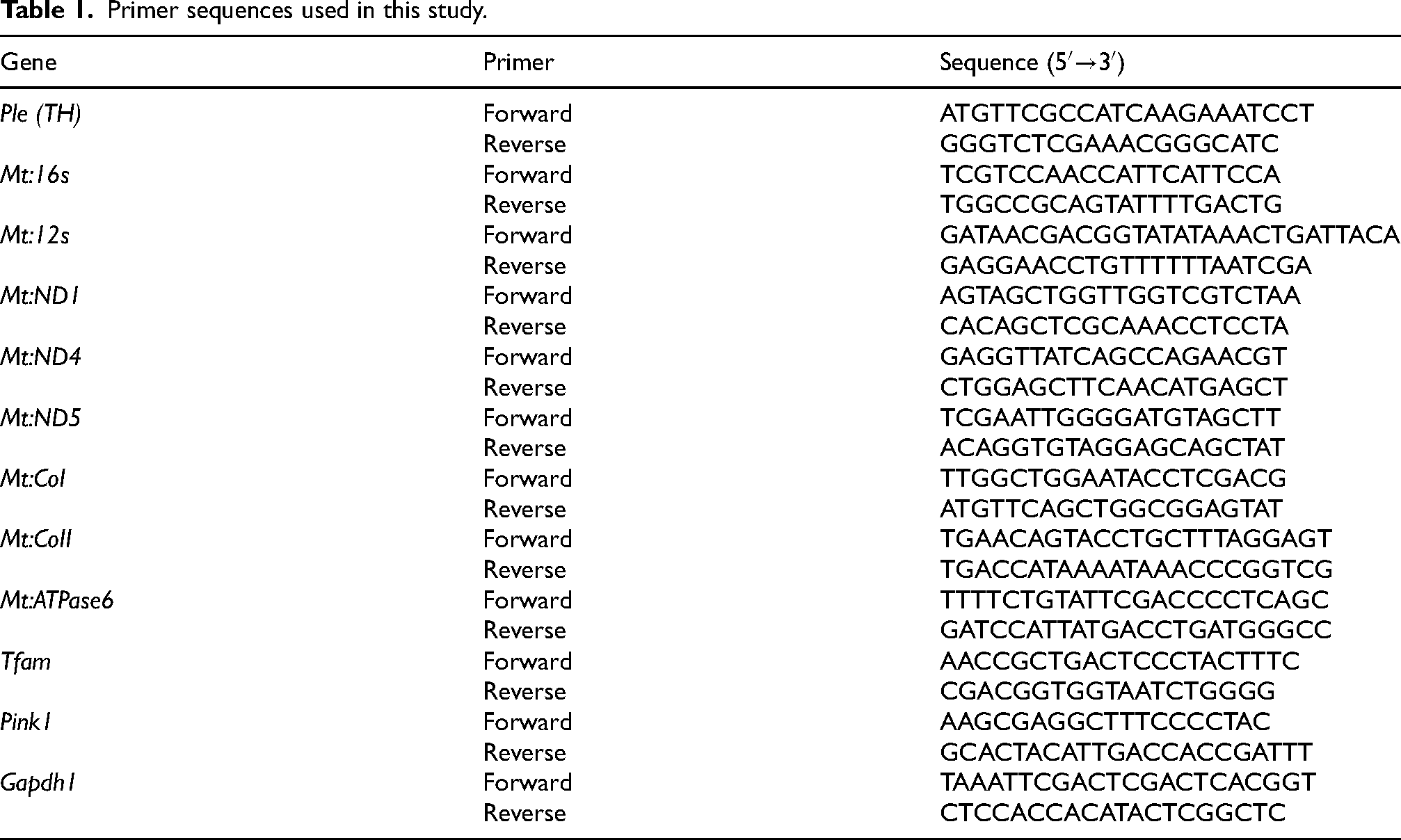
Total RNA was extracted from the adult male heads of 2-day-old flies (n = 8–10). 13 The heads were isolated by placing the e-tubes containing flies in liquid nitrogen for quick-freezing, followed by vortexing for 10 s. The reagents and subsequent procedures were based on previous studies. 8 RNA (4.5 ng) was reverse-transcribed to synthesize cDNA. Gapdh1 Ct values were used for normalization. Primers for qRT-PCR were designed using the FlyPrimerBank (https://www.flyrnai.org/flyprimerbank). 14 Primers were synthesized by Bioneer (Daejeon, Korea). The primers used in this study are listed in Table 1.
Primer sequences used in this study.
Western blotting
The procedures and reagents used for western blotting have been described previously. 8 Total proteins were extracted from the adult male heads of 2-day-old flies (n = 8–10). The primary antibodies used were rabbit anti-TH antibody (1:500, Novus Biologicals, Centennial, CO, USA), rabbit anti-pAKT (Ser473) antibody (1:500, Cell Signaling Technology, Boston, MA), rabbit anti-AKT antibody (1:1000, Cell Signaling Technology, Boston, MA), mouse anti-Actin antibody (1:10000, Abcam, Cambridge, UK). The primary antibodies were incubated at 4 °C for 24 h. The secondary antibodies used were anti-rabbit IgG (1:1000, Cell Signaling Technology, Boston, MA) and anti-mouse IgG (1:10000, Cell Signaling Technology, Boston, MA). The secondary antibodies were incubated at room temperature for 2 h.
Immunofluorescence staining
The procedures and reagents used for immunofluorescence staining have been described previously. 9 The brains of adult male flies were dissected in 0.2% PBSTx solution. The primary antibody (anti-TH, 1:500) was incubated for > 48 h at 4°C. The secondary antibody (Alexa Fluor 488, anti-rabbit, 1:1000) was incubated at room temperature for 2 h. Observations were performed at 200x magnification using an LSM880 laser scanning confocal microscope (Carl Zeiss, Jena, Germany). All images were processed using Zeiss Blue (Carl Zeiss, Jena, Germany) and ImageJ (National Institutes of Health, Bethesda, MD, USA) for image analysis and quantification.
Life span assay
Each genotype fly was collected during eclosion and divided into vials of 10 flies per vial. Flies were transferred to fresh vials every 3–4 days, and the number of dead flies was counted. Survival rates were calculated based on the total population.
Climbing ability
The procedures and reagents used for climbing have been described previously.8,9,15 The observations were performed using an iPhone SE 2 (Apple, Cupertino, CA, USA). The assay was conducted only at specified times (5–7 PM) to determine the circadian rhythm of Drosophila.
Brain histology
The procedures and reagents used for paraffin embedding have been described previously.9,16 Observations were performed at 100x magnification using an Axio Vision microscope (Carl Zeiss, Jena, Germany).
Detection of ROS in brain tissue
The procedures and reagents used for the detection of ROS using DHE have been described previously.9,17 Observations were performed immediately at 200x magnification using an LSM880 laser scanning confocal microscope (Carl Zeiss, Jena, Germany). Images were obtained as Z-series at equal intervals (1 μm) throughout the entire brain. Z-series images were merged and the fluorescence intensity was measured using Zen blue software.
Mitochondrial ROS detection using MitoSOX Red
To specifically detect mitochondrial superoxide, brains were dissected in Schneider's Drosophila medium and incubated with MitoSOX Red mitochondrial superoxide indicator (Invitrogen, Carlsbad, CA, USA) at 30 μM for 5 min at room temperature in the dark, followed by washes three times with Schneider's Drosophila medium. Samples were mounted in Fluoromount-G. Images were obtained at 200x magnification using an LSM880 confocal microscope (Carl Zeiss, Jena, Germany). Z-stack images (1 μm intervals) were acquired and projected using the maximum intensity method. Quantification of fluorescence intensity was performed using ImageJ. A fixed-size rectangular ROI (224 × 224 pixels) was manually positioned over the PPL1 cluster in all samples to ensure consistent anatomical comparison.
Cell death analysis
To detect dying cells in the adult brain, a TUNEL assay was conducted using TMR Red. For TMR Red staining, brains were dissected in 0.2% PBSTx and fixed in 4% paraformaldehyde. Subsequent procedures and reagents used are as described previously.8,18 Following mounting, observations were performed immediately at 200x magnification using an LSM880 laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
Statistical analysis
All statistical analyses were performed using GraphPad Prism v9.02 and imageJ software. The unpaired t-test was used to compare two groups and ordinary one-way ANOVA was used to compare more than three groups to determine statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Knockdown of Pss gene in cortex glia decreases lifespan and motor ability
The common characteristics of patients with PD include reduced lifespan and impaired motor ability. In our previous study, we demonstrated evidence of neurodegeneration following either knockdown or overexpression of Pss in pan-glial cells. Among the various types of glia, knockdown of Pss in cortex glia had the most significant impact on lifespan. In this study, we examined the expression pattern of NP2222-GAL4, which targets cortex glia. By crossing NP2222-GAL4 flies with UAS-mCherry.nls and performing double immunostaining with anti-repo, we confirmed that the expression was restricted to glial cells. Notably, we observed prominent expression near the protocerebral posterior lateral 1 (PPL1) region, one of the dopaminergic neuron clusters (Figure 1D, E). To improve clarity, we now define PPL1 in the beginning of the Results section as a cluster of tyrosine hydroxylase (TH)-positive dopaminergic neurons known to be selectively vulnerable in Drosophila PD models. Next, we investigated these two characteristics in flies with regulated Pss gene expression in the cortex glia (Figure 1A, B). The T50 value, which is the time at which half of the individuals are alive, was approximately 46 days for females and 37.5 days for males in the control group. In contrast, the fly group in which the Pss gene was knocked down in cortex glia showed a substantially decreased lifespan. The T50 values for females and males were 5 and 2 days, respectively. However, the group in which the Pss gene was overexpressed in cortex glia did not show a statistically significant difference from the control group. The T50 value was 46 days for females and 39 days for males in flies in which the Pss gene was overexpressed. To measure the motor ability, we performed a climbing assay using male flies of the same age (Figure 1C). The average height of each fly in 10 s was 7.97 cm for the control group, but only 2.026 cm for flies with Pss gene knockdown in the cortex glia. Notably, flies with Pss overexpression in the cortex glia increased by an average of 11.92 cm, which was higher than that in the control group.
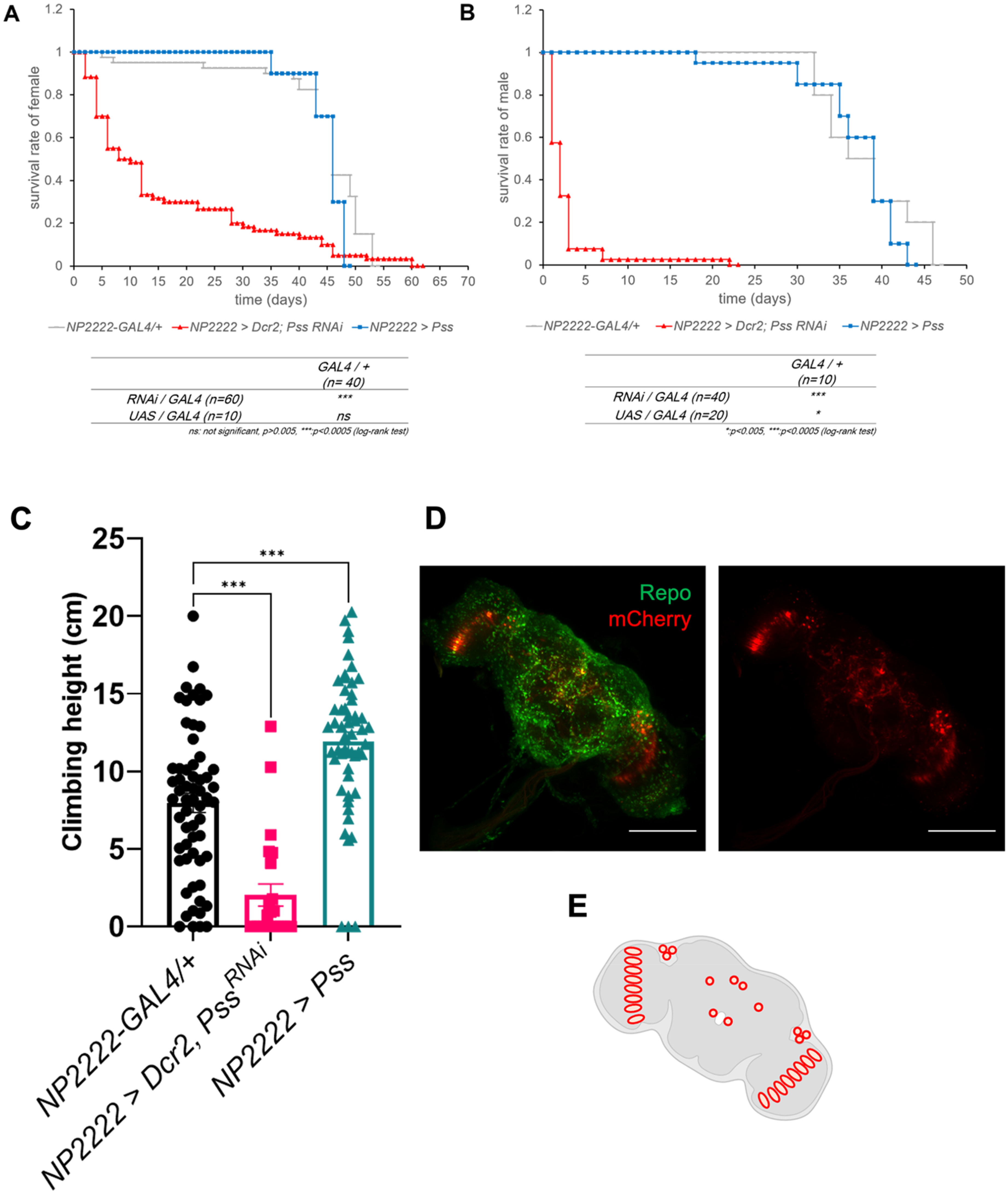
Knockdown of Pss in cortex glia leads to decreased lifespan and impaired motor ability. (A) Lifespan analysis of female Drosophila cultured at 29°C. NP2222-GAL4/+ (n = 40), NP2222 > Dcr2, PssRNAi (n = 60), NP2222 > Pss (n = 10). Log rank test: *p < 0.005, ***p < 0.0005. (B) Lifespan analysis of male Drosophila cultured at 29°C. NP2222-GAL4/+ (n = 10), NP2222 > Dcr2, PssRNAi (n = 40), NP2222 > Pss (n = 20). Log rank test: *p < 0.005, ***p < 0.0005. (C) Knockdown of Pss in cortex glia leads to locomotion defect. Negative geotaxis assay was performed using 2-day-old flies. NP2222-GAL4/+ (n = 60), NP2222 > Dcr2, PssRNAi (n = 30), NP2222 > Pss (n = 60). t-test: *p < 0.05, **p < 0.01, ***p < 0.005. (D) Representative confocal image showing of NP2222-GAL4-driven expression of UAS- mCherry.nls. Green is anti-repo expressed in glia. Scale bar:
Knockdown of pss gene in cortex glia induces cell death in a head
We observed that various neurodegenerative phenotypes resulting from the knockdown of Pss gene in cortex glia prevented the CNS from maintaining normal function. Therefore, to histologically analyze the brain morphology, we embedded the heads of 1-day-old flies in paraffin and stained them with hematoxylin and eosin. We observed more vacuoles and empty spaces in the cortex of the brain with Pss gene knockdown in the cortex glia (Figure 2A). We also performed a TUNEL assay, which labels fragmented DNA, to determine whether the bigger holes were due to cell death (Figure 2B). The results showed a higher distribution of TUNEL-positive cells in the Pss gene-knockdown flies than in the control group, with an approximately 1.5 times higher intensity. Although the precise cell types undergoing cell death could not be determined, future co-staining with glial and neuronal markers will help distinguish the affected populations. Collectively, these results suggested that the reduced lifespan and impaired motor ability observed in Pss knockdown flies were caused by abnormal CNS structures owing to increased cell death.
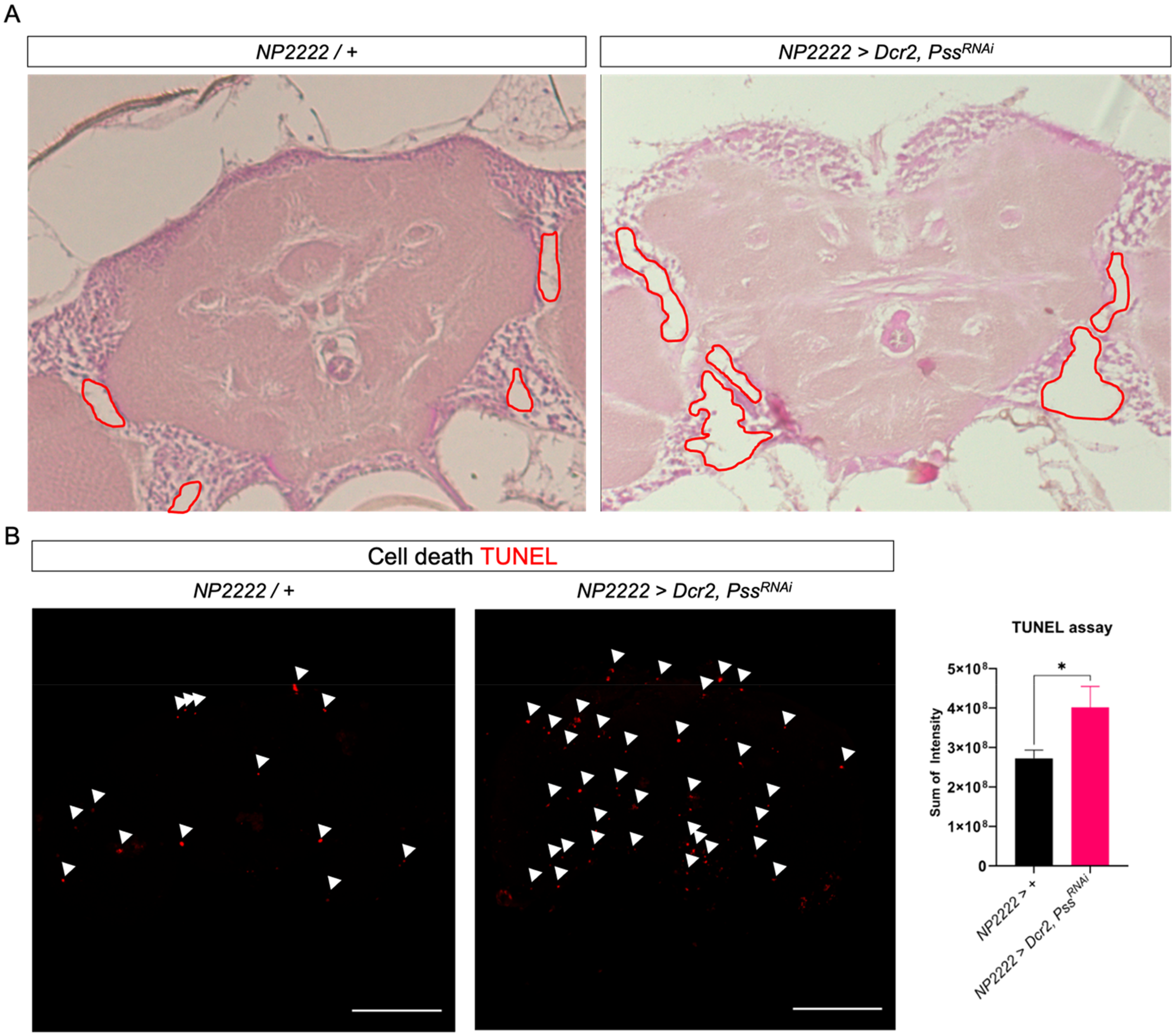
Pss knockdown individuals exhibit abnormal retinal structure and induce cell death. (A) Brain section of 2-day-old flies. Compared to the straight elongated controls, numerous gaps were observed in Pss KD mutant individuals. (B) Brain of 2-day-old flies. TUNEL staining was used to detect cell death. Intensity of TUNEL staining (t-test: *p < 0.05, **p < 0.01, ***p < 0.005). Scale bar:
Knockdown of Pss gene in cortex glia induces dopaminergic neuronal cell death
We observed various neurodegenerative phenotypes resulting from knockdown of the Pss gene in cortex glia, such as reduced motor ability and cell death, in PD. Therefore, we investigated whether the most representative feature of PD, a reduction in dopaminergic neurons, was present. Dopamine is produced by the l-DOPA-synthesizing enzyme, TH, in dopaminergic neurons. Therefore, we examined TH expression to determine the presence of dopaminergic neuronal cells. After homogenizing the heads of 1-day-old flies and analyzing their expression through qRT-PCR and western blotting, we found that RNA levels were significantly reduced by 30% in flies with Pss knockdown in the cortex glia compared to those in the control group, and protein levels were also reduced, as confirmed using western blotting (Figure 3A, B). To verify the reduction in the number of dopaminergic neurons, we performed immunofluorescence staining using anti-TH (Figure 3C). The number of dopaminergic neurons was markedly reduced in both the PPL1 and PPL2 clusters in flies with Pss knockdown in the cortex glia, with an average decrease of 2.917 and 2 neurons, respectively, compared with the control group. Of note, flies with Pss overexpression in cortex glia showed a statistically significant reduction of 3.65 neurons on average in the PPL1 cluster, but did not show differences in other dopaminergic cell clusters. Furthermore, to determine whether the observed cell loss was associated with apoptosis, we performed double staining with anti-TH and TUNEL in fly brains (Figure 3D). Apoptotic signals were detected in close proximity to TH-positive dopaminergic neurons in the PPL1 cluster of flies with Pss knockdown, indicating that dopaminergic neuron loss may be caused by apoptosis.
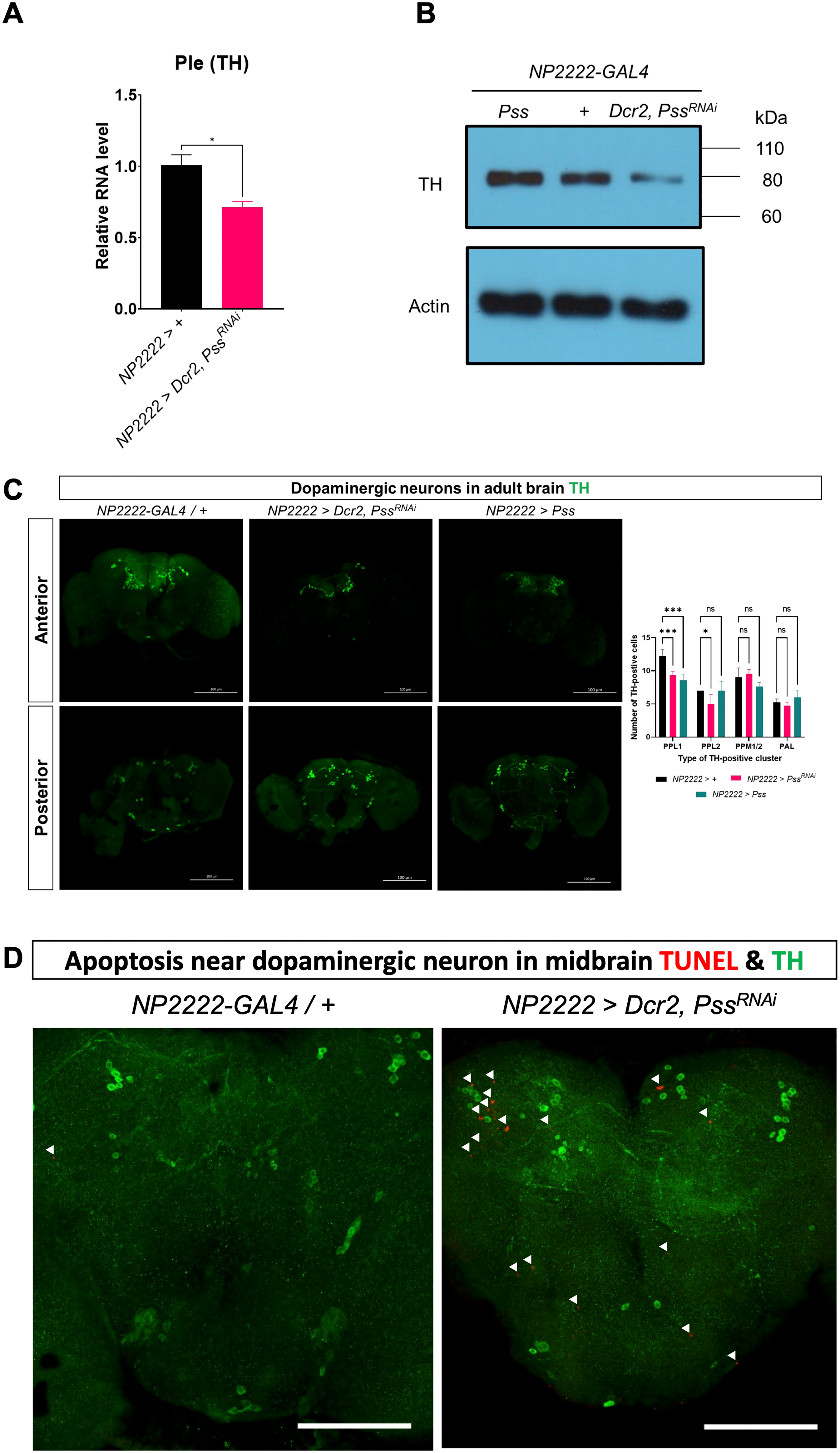
Pss knockdown induces a decrease in TH expression and TH-positive cell count. (A) Ple (TH) gene transcription decreased in NP2222 > Dcr2, PssRNAi individuals (n = 3). (B) TH protein decreased in NP2222 > Dcr2, PssRNAi individuals (n = 3). (C) Cluster-specific counts of anti-TH (dopaminergic neurons) decreased in NP2222 > Dcr2, PssRNAi individuals (n = 5) (arrow head; TUNEL). Scale bar:
Knockdown of Pss gene in cortex glia results in mitochondrial dysfunction
We then investigated whether the Parkinson's disease-related phenotypes resulting from the knockdown of Pss gene in cortex glia were associated with mitochondrial abnormalities, one of the main causes of PD. Mitochondrial DNA (mtDNA) is damaged when the mitochondria are damaged by ROS and other factors. If damaged mitochondria are not removed, they can lead to cell death, ultimately causing neurodegenerative diseases, such as PD. Therefore, we examined the expression of various mtDNA to confirm mitochondrial dysfunction (Figure 4A). Mitochondrial rRNA expression increased by a statistically significant 25.5% in the Pss knockdown flies compared to control flies. However, the expression of ATPase6, a protein involved in ATP production in the mitochondrial electron transport chain complex V, decreased by a statistically significant 75%. We then examined the abnormalities in the expression of the Tfam gene, which regulates mtDNA expression at the upper level (Figure 4B). Tfam RNA expression decreased by a statistically significant 45% in the Pss knockdown flies compared to control flies. These results suggest that the inhibition of Pss expression in cortex glia reduces Tfam expression, leading to decreased mtDNA expression and mitochondrial dysfunction. We also examined the expression of Pink1 gene, associated with mitophagy (Figure 4C). Pink1 RNA expression was significantly increased by 50% in Pss knock-downed flies compared to the control group. Finally, we examined ROS levels because mitochondrial dysfunction subsequently increases ROS levels (Figure 4D). Flies with specific knockdown of Pss expression in cortex glia showed significantly higher levels of ROS than the control group, and ROS was present at a high concentration in dopaminergic neurons. To confirm that the observed ROS originated from mitochondrial dysregulation in cortex glia, we performed MitoSOX staining following Pss knockdown. Maximum intensity projections of z-stack images were normalized and pseudo-colored for visualization (Figure 5A). Quantification of either mean fluorescence intensity or the thresholded sum intensity of MitoSOX-positive puncta restricted to a fixed-size region of interest (ROI) positioned over the PPL1 cluster—a region containing dopaminergic neurons—both measurements revealed a statistically significant increase in mitochondrial ROS in Pss knockdown samples compared to controls (p < 0.01, **) (Figure 5B, 5C). A fixed-size rectangular ROI (224 × 224 pixels) was consistently positioned over the PPL1 cluster to ensure anatomical precision and inter-sample comparability. These findings are consistent with our DHE-based measurements of cytoplasmic ROS and suggest that Pss may regulate both cytoplasmic and mitochondrial oxidative tone in cortex glia, although the sensitivity of the assays may differ. We did not directly examine non-dopaminergic neurons, but the fact that MitoSOX signal was mainly increased in dopaminergic neuron areas suggests some level of cell-type specificity. These findings suggest that the knockdown of Pss expression in cortex glia suppresses Tfam expression and reduces mtDNA expression, impairing normal mitochondrial function.
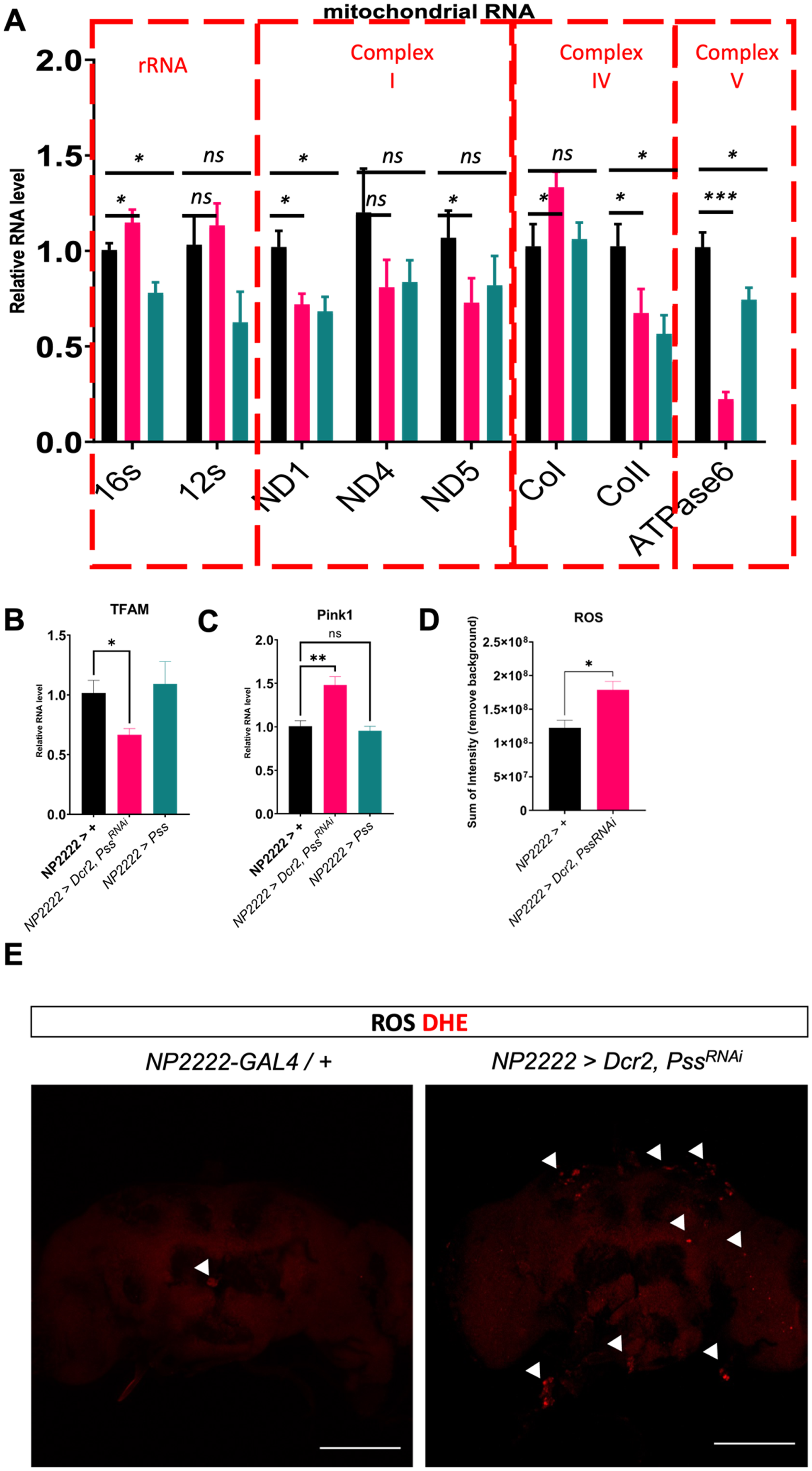
Knockdown of Pss in cortex glia leads to mitochondrial abnormalities. (A) Differences in expression levels of genes on mitochondrial DNA shown using qRT-PCR. (B) Differences in expression levels of TFAM, which regulates genes on mitochondrial DNA, shown using qRT-PCR. (C) Differences in expression levels of Pink1, which regulates mitophagy, shown using qRT-PCR. (D) Quantification of ROS detection in the brain using DHE. Four samples were measured for quantification (arrow head). Scale bar:
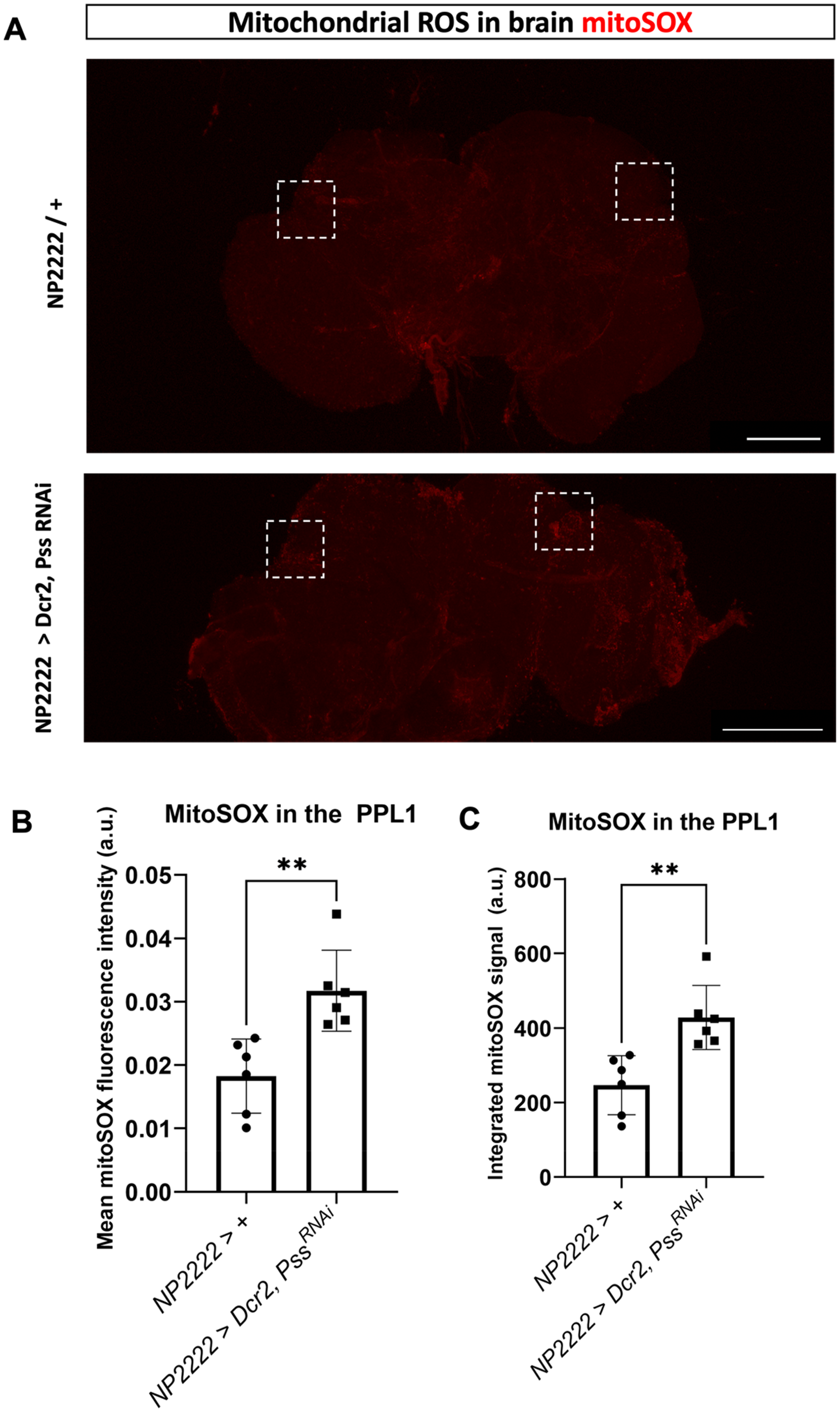
Mitochondrial ROS levels in cortex glia following Pss knockdown.
Pss knockdown suppresses AKT signaling in cortex glia
Since AKT is known to activate Tfam expression, and PS has been reported to promote AKT signaling, we investigated whether Pss knockdown affects AKT signaling in the brain. We performed Western blotting using adult fly head lysates. In cortex glia-specific Pss knockdown flies, phosphorylated AKT levels were strongly reduced, while total AKT protein levels remained unchanged (Figure 6A, B). Actin levels confirmed equal loading (Figure 6C). These results indicate that Pss knockdown suppresses AKT pathway activation without altering total AKT expression.
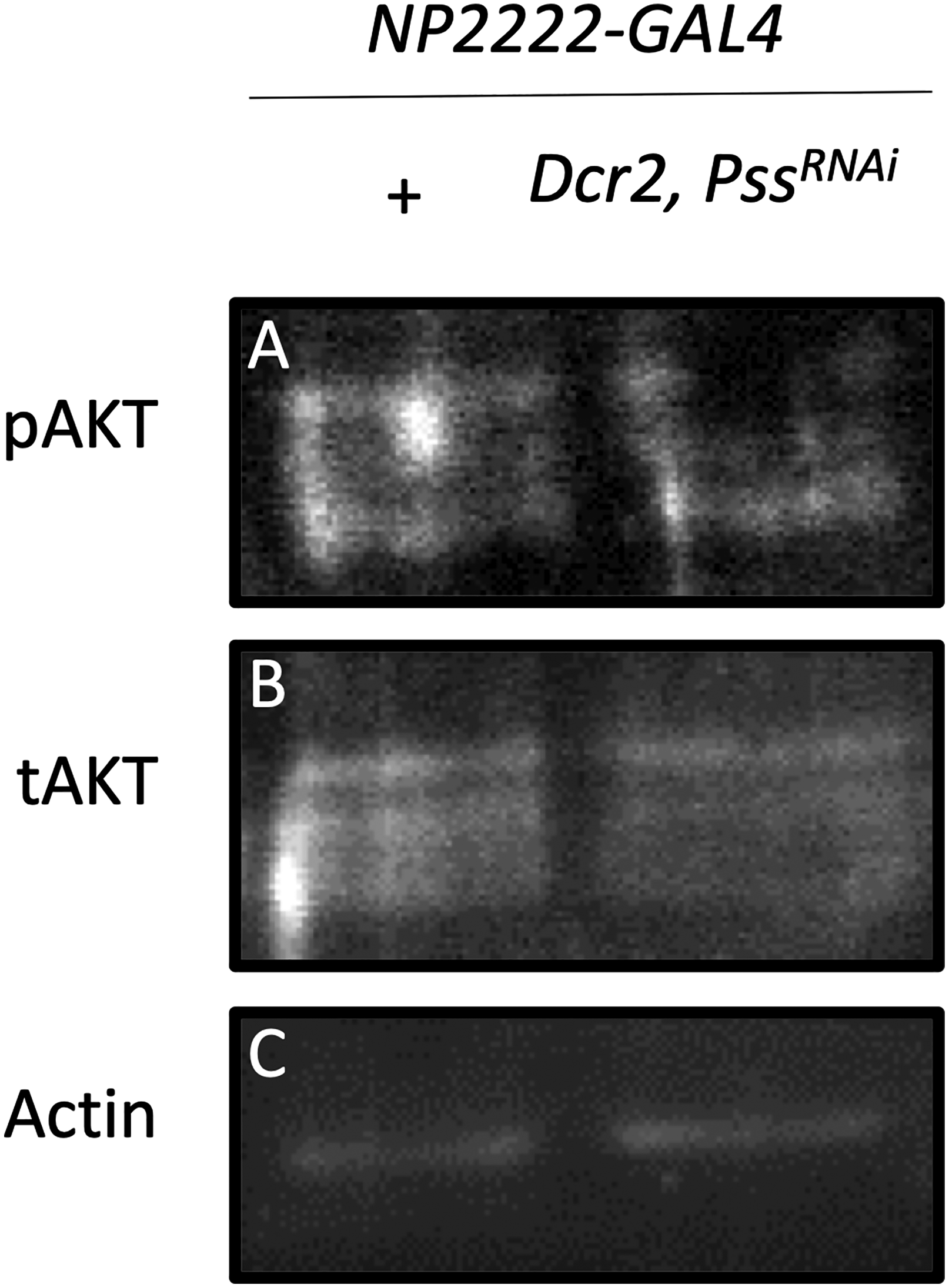
Western blot analysis of AKT signaling. (A) Phosphorylated AKT (pAKT); (B) Total AKT (tAKT); (C) Actin as a loading control. Pss knockdown in cortex glia reduces the level of pAKT, while tAKT levels remain unchanged.
Discussion
Despite extensive research on PD, the exact cellular and molecular mechanisms underlying the disease remain poorly understood. 1 In particular, phospholipid dysregulation has recently gained increasing attention as a possible contributor to PD. 19 Notably, PS, a negatively charged phospholipid enriched in neuronal membranes, has been implicated in mitochondrial stability, apoptotic signaling, and membrane-based kinase activity. Several studies have reported that PS homeostasis is disrupted in PD patients. The substantia nigra, a key region affected in PD, has been shown to exhibit reduced expression of PTDSS1, one of the enzyme responsible for PS synthesis in mammals, based on transcriptomic profiling. 20 In contrast, increased PS levels have been found in the plasma and frontal cortex of early-stage PD patients.21,22 Furthermore, clinical studies have suggested that oral PS supplementation may improve motor symptoms in PD patients. 23 These findings highlight PS as a potentially critical regulator in PD pathogenesis, yet the causal mechanisms remain poorly defined. To investigate this further, we used a Drosophila model to manipulate Pss—the homolog of PTDSS1—in cortex glia, and examined its impact on mitochondrial function, glial health, and dopaminergic neuron integrity.
We previously showed that knockdown of Pss in glial cells leads to neurodegeneration in Drosophila, and also found that muscle-specific knockdown results in progressive muscle atrophy.8,9 These phenotypes became more severe over time, similar to how symptoms develop in PD. To further investigate PD-related phenotypes, we knocked down Pss specifically in cortex glia, a subtype of glial cells in Drosophila. Flies with Pss knockdown in cortex glia exhibited shortened lifespan, impaired motor ability, mitochondrial dysfunction, and reduced number of dopaminergic neurons—all hallmark features of PD.8,9,24 Glial cells contribute to inflammation, metabolism, regeneration, and myelination, supporting the proper development and function of the nervous system. 25 Disruption of glial function impairs neural activity and ultimately contributes to the development of neurological diseases. 26 Glia are divided into functional subtypes, and abnormalities in astrocytes and microglia, in particular, have been implicated in mammalian models of PD.25,27–33 Because Drosophila cortex glia exhibit both astrocyte-like and microglia-like functions in the CNS, their dysfunction is thought to provide insights into the pathogenesis of PD.28,29,34,35
PS synthesized in the endoplasmic reticulum is trafficked to both the plasma membrane and mitochondria. 36 At the membrane, PS facilitates AKT activation through phosphorylation. 37 In mitochondria, PS serves as a precursor of PE. A reduction in PS levels therefore leads to decreased PE production, impairing mitochondrial structure and function.8,9,24 Consistent with previous studies in muscle and salivary glands, our results also showed that Pss knockdown in cortex glia impairs AKT signaling. Specifically, we observed a clear reduction in phosphorylated AKT, while total AKT protein levels were unaffected. This supports the notion that PS is required for proper AKT activation, possibly through effects on membrane localization or upstream signaling components. Impaired AKT signaling may contribute to downstream events such as mitochondrial dysfunction or transcriptional changes, including mitochondrial transcription factor A downregulation.38–40 As a result, the transcription of multiple mitochondrial genes—such as ND1 and ND5 (complex I), CoII (complex IV), and ATPase6 (complex V)—was suppressed, compromising mitochondrial function. This mitochondrial dysfunction triggered a compensatory increase in Pink1 expression, indicative of impaired mitophagy, and ultimately contributed to elevated mitochondrial ROS levels. In addition to DHE-based detection of cytoplasmic ROS, we directly assessed mitochondrial ROS using MitoSOX staining, which selectively detects mitochondrial superoxide. Targeted quantification within the PPL1 region—a dopaminergic neuron cluster—showed a significant increase in MitoSOX signal. This finding is consistent with the DHE results and supports the idea that mitochondrial redox homeostasis is disrupted following Pss knockdown. Taken together, our findings demonstrate that glial PS metabolism is essential for mitochondrial homeostasis and dopaminergic neuron survival, and that its dysregulation may represent a previously underappreciated contributor to PD pathogenesis. Nevertheless, the translational relevance of this model is inherently limited, and validation in mammalian systems will be essential to confirm the broader applicability of our findings.
Footnotes
Acknowledgements
We thank Ye-Jin Park for giving constructive advice on this project. We also appreciate the technical support provided by Korea Basic Science Institute, Seoul Center, Korea.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Research Foundation of Korea grants funded by the Korean government (MSIT; Nos. 2019R1F1A106177 and RS-2023-00253615), the BK21 FOUR Program through the Center for Science Education in the Infosphere, and Korea Basic Science Institute under the R&D program (C523400).
Data availability statement
Not applicable. No datasets were generated or analyzed during the current study.